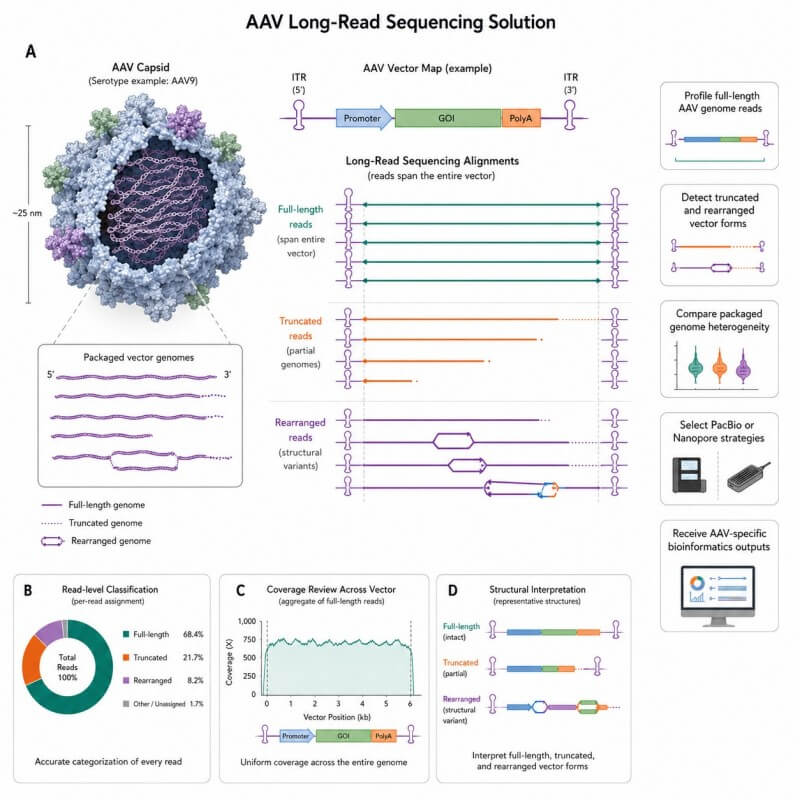
Why AAV Vector Genome Structure Needs More Than Short Reads
Short-read sequencing is useful for local sequence review, coverage analysis, and variant-level evidence. However, many AAV research questions are not only local sequence questions. They are structure questions.
An AAV preparation may contain reads that match the expected construct, but it may also include partial genomes, truncated forms, rearranged molecules, concatemers, or other unexpected packaged DNA patterns. When the evidence is split into short fragments, these larger structures can be difficult to interpret with confidence.
Long-read sequencing gives your team a broader view of the vector genome. Reads that span longer regions can help show how different parts of the vector are connected. This is especially useful when your study needs to understand whether packaged genomes are full-length, where truncation patterns occur, or whether unexpected structures are present.
For many AAV vector research teams, the key question is not only, "Is the designed sequence present?" A more useful question is: what genome forms are actually represented in the packaged vector population?
Our solution is designed to answer that question through long-read sequencing, vector-aware mapping, read-level classification, structure annotation, and clear reporting.