ATAC-Seq FAQs
1. What type of controls are necessary for ATAC-Seq experiments?
In the realm of ATAC-Seq experiments, the incorporation of diverse controls is imperative to uphold precision and dependability. The utilization of an input DNA control serves the purpose of normalizing potential biases in DNA preparation and sequencing processes. Technical replicates play a crucial role in confirming the reproducibility and reliability of the outcomes. The inclusion of positive controls, representing established open chromatin regions, acts as a mechanism to authenticate the protocol. On the other hand, negative controls, indicative of anticipated closed chromatin regions, aid in the detection and mitigation of background noise.
2. How does ATAC-Seq compare to other chromatin accessibility assays like DNase-Seq and FAIRE-Seq?
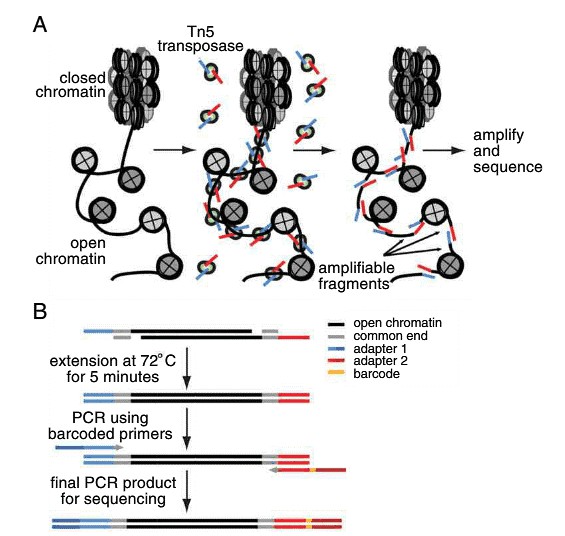
DNase-Seq, ATAC-Seq, and FAIRE-Seq are all techniques utilized for studying open chromatin regions. DNase-Seq employs the use of DNase I endonuclease to identify accessible chromatin regions, FAIRE-Seq involves sonication followed by phenol-chloroform enrichment, and ATAC-Seq utilizes Tn5 transposase for enrichment and amplification. The high activity of Tn5 transposase makes ATAC-Seq a straightforward, efficient method requiring only 500-50,000 cells. The sensitivity and specificity of ATAC-Seq are comparable to DNase-Seq and superior to FAIRE-Seq.
3. What techniques are commonly combined with ATAC-seq for research?
ATAC-seq + RNA-seq: Generally, RNA-seq is performed prior to ATAC-seq to identify differentially expressed genes and enriched pathways, which suggest correlations. To determine which factors regulate these target genes, motif analysis via ATAC-seq can identify potential regulatory elements. Subsequent validation can be carried out through additional experiments or ChIP-seq.
ATAC-seq + Hi-C: For studies investigating the effects of higher-order chromatin structures on biological processes, ATAC-seq is often used alongside Hi-C. Hi-C provides information on higher-order structures like compartment A/B, TADs, and loops, which indicate correlations. ATAC-seq can further identify promoters, enhancers, and other elements, elucidating how these structures influence gene expression.
ATAC-seq + Histone Modifications: While ATAC-seq can predict the accessibility of specific sites and potential transcription factor binding, it does not reveal whether these factors promote or inhibit gene expression. Combining ATAC-seq with histone modification data (e.g., H3K27ac for activation or H3K27me3 for repression) can provide a more comprehensive understanding, making the data more robust and reliable.