PacBio SMRT Sequencing FAQ
1. What makes PacBio HiFi different from other long-read sequencing?
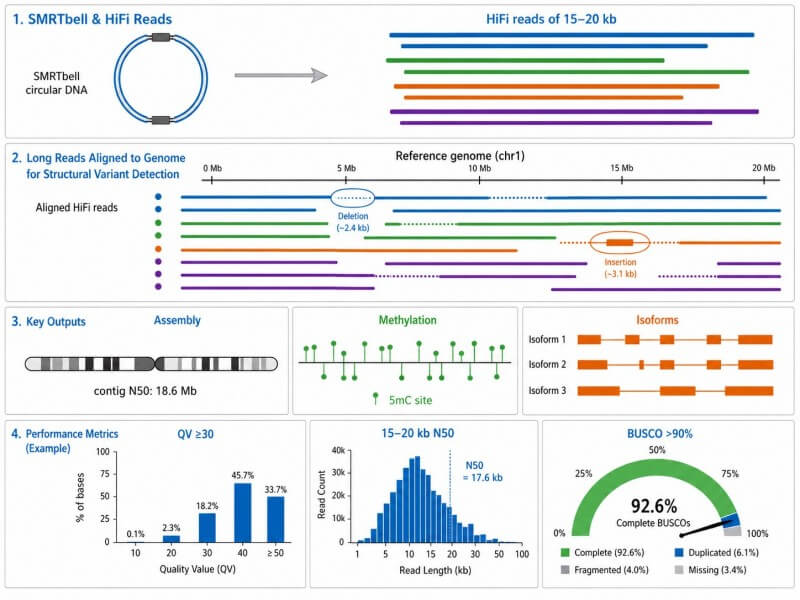
HiFi is the only long-read technology that delivers single-molecule QV ≥30 accuracy. It achieves this through circular consensus sequencing — each molecule is read multiple times and the passes are combined. By contrast, nanopore raw reads have lower per-base accuracy and typically require consensus or polishing steps. HiFi also detects 5mC methylation from polymerase kinetics in the same run, with no additional chemistry required.
2. How does HiFi accuracy compare to Illumina?
HiFi reads match Illumina's per-base accuracy (≥99.9%) while being 50–100× longer. This means you can call SNVs with the same confidence as short reads — but also detect structural variants, phase haplotypes, and assemble genomes without gaps. HiFi is not a trade-off between accuracy and length; it gives you both.
3. Can I detect methylation from my PacBio run?
Yes — and you don't need to do anything extra. PacBio polymerases slow down at methylated bases, producing kinetic signatures that the basecaller interprets as 5mC calls. These calls are generated during standard CCS analysis. No bisulfite conversion, no separate library, no parallel sequencing run. You get variants and methylation from one SMRT Cell.
4. What is Iso-Seq and when should I use it instead of short-read RNA-seq?
Iso-Seq (now also available as Kinnex for higher throughput) sequences full-length cDNA molecules from transcription start site to polyA tail in single reads. Use it when you need to identify complete isoform structures, detect fusion transcripts, or quantify isoform usage — tasks where short-read RNA-seq guesses at isoform assembly. Kinnex concatenates multiple transcripts per read, increasing throughput per SMRT Cell ~5-fold.
5. How much DNA do I need?
Standard WGS: ≥1–5 μg HMW gDNA at ≥30 kb modal length. T2T projects: ≥3–5 μg at ≥50 kb. Iso-Seq: ≥500 ng–1 μg total RNA at RIN ≥8. Lower inputs may be possible — contact us with your sample constraints.
6. How long does a project take?
Project timelines depend on library preparation, sequencing configuration, and bioinformatics scope. We provide a detailed timeline during project consultation based on your specific requirements.
7. What instruments do you use?
PacBio Sequel II/IIe and Revio systems, SMRT Cell 8M and 25M formats. All runs are monitored by experienced technicians with SOP-driven QC gates.
8. Can I send my own SMRTbell libraries?
Yes. Our Pre-Made Library Sequencing service accepts customer-prepared libraries. We QC on arrival, load onto the appropriate SMRT Cell, and deliver CCS data plus optional bioinformatics.
9. How is this different from the PacBio services I can get elsewhere?
Three differences: (1) We optimize each SMRT Cell for multiple data types — you get variants AND methylation AND assembly metrics from the same run. (2) Every project includes a publication-ready QC report with metrics reviewers expect. (3) We provide cross-platform guidance — if a hybrid HiFi + ONT or HiFi + Illumina design would serve your question better, we'll recommend it.