CD Genomics offers MeDIP‑seq for genome‑wide methylation profiling and hMeDIP‑seq to selectively capture 5‑hydroxymethylcytosine (5hmC), enabling parallel analysis of multiple cytosine modifications.
Introduction to MeDIP seq and hMeDIP seq
DNA methylation refers to the postreplicative maintenance or de novo addition of a methyl group to the carbon-5 position of the cytosine pyrimidine ring by DNA methyltransferases. In mammals, DNA methylation typically occurs in CG (mCG) and non-CG (mCHG or mCHH, collectively referred to as mC) contexts. mCG alone accounts for approximately 60–80% of all CG dinucleotides. In addition to 5-methylcytosine (5mC), several cytosine modifications have been identified, including 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine. Among them, 5hmC is structurally similar to 5mC but much less abundant, and is generated through the oxidation of 5mC by the TET family of dioxygenases.
DNA methylation and hydroxymethylation are both crucial epigenetic marks implicated in gene regulation, embryonic development, cellular differentiation, and genome stability. Genome-wide analysis of both 5mC and 5hmC provides critical insight into epigenetic dynamics, particularly in developmental and stress response studies.
MeDIP‑seq couples immunoprecipitation with high-throughput sequencing to enrich and profile 5mC across the genome. It utilizes anti‑5mC antibodies to selectively capture DNA fragments containing methylated cytosines (in mCG and mCH contexts). These enriched fragments are sequenced, and their read distribution can be used to estimate relative methylation levels.
To distinguish and specifically profile 5hmC, CD Genomics also offers hMeDIP‑seq, which employs 5hmC‑specific antibodies to enrich hydroxymethylated DNA fragments. This complementary approach allows researchers to separately profile 5mC and 5hmC modifications in parallel, enhancing the resolution of epigenetic mapping across diverse sample types. Both MeDIP‑seq and hMeDIP‑seq are non-destructive, conversion-free methods that do not rely on bisulfite treatment or uracil-tolerant polymerases, and are compatible with low-input samples.
Advantages of MeDIP-Seq / hMeDIP-Seq
Can target mC, mCG, or hmC
Whole genome or any regions of interest
Near-unbiased and hypothesis
Epigenetic biomarker discovery
Single-nucleotide resolution and cost-efficiency
Low input DNA requirement
Applications of MeDIP-Seq / hMeDIP-Seq
Epigenetic Heterogeneity
Environment and Epigenetics
Genetic Expression Regulation
Disease Research
Genetic Imprinting
Embryonic Development
Detection of DNA methylation-enriched regions in disease samples and inference of candidate gene sets whose expression is suppressed in the samples.
Monitoring the dynamic DNA methylation patterns at different stages of disease occurrence and development, particularly in lesion sites, to screen for epigenetic markers that help define the extent of disease progression.
Comparative analysis of the differences in signal and location distribution of DNA methylation regions between disease and normal samples, identification of disease-specific DNA methylation regions, and observation of the genes surrounding these regions to narrow down the list of candidate genes related to the disease.
Mapping of hydroxymethylation (5hmC) landscapes in tissues, cell differentiation, or stress-response models using hMeDIP-seq.
MeDIP-Seq / hMeDIP-Seq Workflow
The general workflow for MeDIP sequencing is outlined below. Briefly, the extracted DNA is fragmented, denatured, ligated with adaptor and captured using the antibody directed against 5-methylcytosine (for MeDIP-seq) or 5-hydroxymethylcytosine (for hMeDIP-seq), followed by library preparation and sequencing on Illumina platforms.
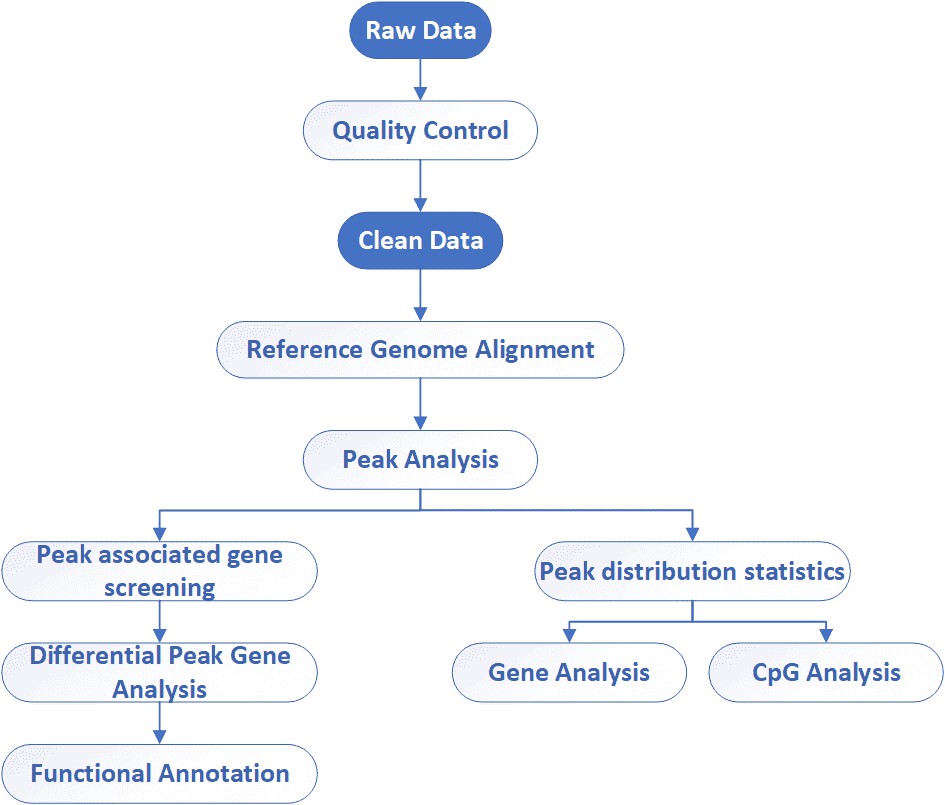
Data Analysis We provide multiple customized bioinformatics analyses:
Alignment against reference genome
Alignment against reference genome
Whole genome region distribution
Peak distribution statistics
Peak associated gene screening
Functional annotation with GO/KEGG analysis
Differential expression level analysis of peak associated genes
Note: Recommended data outputs and analysis contents displayed are for reference only. For detailed information, please contact us with your customized requests.
Analysis Pipeline
Deliverables
The original sequencing data
Experimental results
Data analysis report
Details in MeDIP-Seq / hMeDIP-Seq for your writing (customization)
With professional bioinformatics capability, CD Genomics offers high-quality MeDIP-Seq as an end-to-end, genome-wide epigenetic service to identify differentially methylated regions, and ultimately help to expedite epigenetic research. If you have additional requirements or questions, please feel free to contact us.
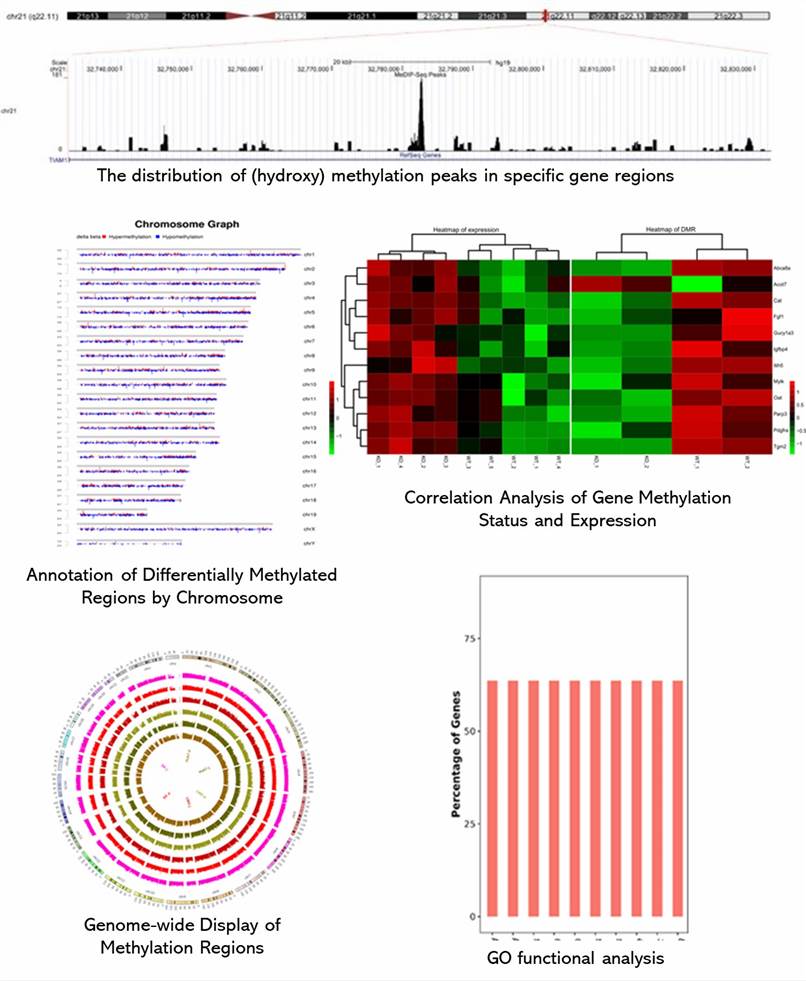
Demo Results
Partial results are shown below:
MeDIP‑Seq / hMeDIP‑Seq FAQs
1. Compared with other methods, what are the advantages of MeDIP sequencing?
Currently, sequencing-based approach for methylome analysis can be classified into bisulfite conversion-based and enrichment-based approaches. Bisulfite conversion-based methods include whole-genome or targeted bisulfite sequencing or reduced representation bisulfite sequencing (RRBS). Although bisulfite conversion-based methods are considered to be the gold standard of DNA methylation analysis at single-base resolution, they cannot distinguish between mC and hmC. Additionally, whole genome bisulfite sequencing is expensive for application on large sample sizes and by smaller research groups. And RRBS can only provide limited genome coverage (5-10%) and is centered on CpG island and promoter regions.
Besides MeDIP sequencing, enrichment-based technologies also include MBD-seq, which uses the methyl-binding protiens MBD2 and MDB3L1, and methylCap-seq that use methyl-binding domain of MECP2 for methyl capture. But MBD-seq and methylCap-seq are restricted to the analysis of mCG and whole-genome protocols often require high concentrations of genomic DNA (more than 1,000 ng). Therefore, MeDIP is a versatile, accurate, and costly method with a low input DNA requirement and is applicable to a wide range of samples and studies.
2. What are the requirements for MeDIP sequencing samples?
MeDIP sequencing samples require reference genome or sequences for alignment, and the assembled results directly affect the accuracy of data analysis. Reference genome from closely related species or assembled results from transcriptome can also be used despite the loss of partial methylation information.
3. What factors can affect the results of MeDIP-seq?
Every process involved in MeDIP-seq may affect the results, especially the immunoprecipitation and PCR. Immunoprecipitation is the process to enrich methylated regions, and reaction conditions may influence the enrichment results. If insufficient DNA is recycled, PCR is used to scale it up, which can biase results. Furthermore, contamination should be avoided throughout the whole course.