Human Mitochondrial DNA (mtDNA) Sequencing
Home
> Human Mitochondrial DNA (mtDNA) Sequencing
Introduction of Human Mitochondrial DNA (mtDNA) Sequencing
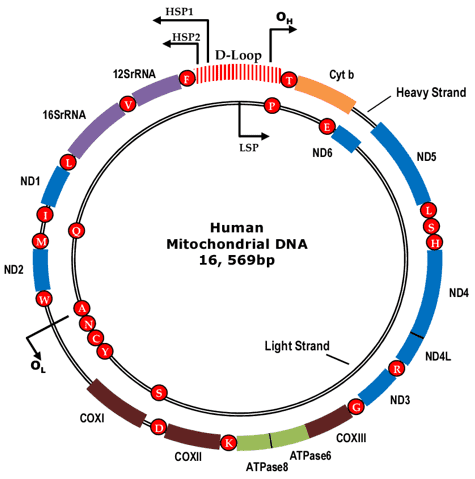
Mitochondria, called "powerhouses of the cell", are vital organelles in human cells. Human mitochondrial DNAs (mtDNA) form double-strand circular molecules that contain about 16,569 DNA base pairs and encode 37 genes (Fig. 1). There are thousands of mtDNA molecules in mtDNA is highly mutable because it is not protected by histones. The mutated mtDNAs coexist with the wide-type ones in a state called heteroplasmy, which has been detected to be associated with a wide range of human diseases, like Autism Spectrum Disorder (ASD), Leber's hereditary optic neuropathy (LHON), breast cancer, Alzheimer's disease, Parkinson's disease and hypertension. Therefore, the mitochondrial genome is a significant object of researches in clinical diagnosis.
N2Jenomics Pvt. Ltd has developed panels that specifically captures the entire human mitochondrial DNA genome. These are multiplex PCR based and probe captured based targeted sequencing approaches which could 100% cover the human mitochondrial genome. Employing this panel, customers no longer need to provide purified mtDNA. mtDNA enrichment is achieved by multiplex PCR amplification or probe capture of the mitochondrial genome. The enriched mtDNA is then subjected to library prep and ultra-deep sequencing.N2Jenomics Pvt. Ltd provides this technology to help customers research on mitochondrial genome-related diseases.
Figure 1. The circular human mitochondrial genome.
Advantages of Our Human Mitochondrial DNA (mtDNA) Sequencing Service
Easier access to initial samples: human gDNA, containing both mtDNA and nuclear DNA
Ultra-deep sequencing: >1000x coverage, with Illumina PE150 platform
High accuracy: 100% amplicon or probe coverage of all regions of the mitochondrial genome
Lowest price: which is dependent on the sample-size, please contact us for a quotation
Fastest turnaround time: 3-4 weeks
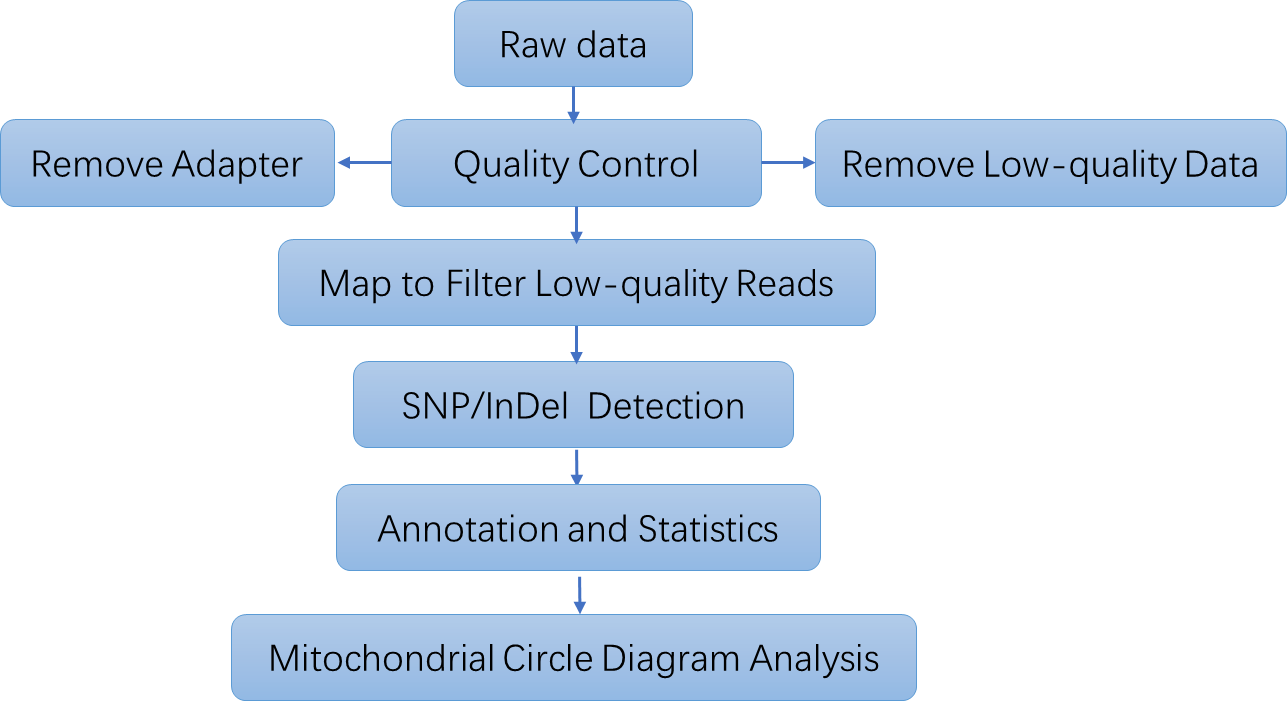
Human Mitochondrial DNA (mtDNA) Sequencing Workflow
Note: Sample amounts are listed for reference only. For detailed information, please contact us with your customized requests.
Click
Sequencing Strategies
Illumina platforms
Capture efficiency: 60%~80%
Bioinformatics Analysis Standard human mtDNA sequencing bioinformatics service includes
Raw data QC
Alignment to the mitochondrial reference genome
Variant (SNP/indel) calling
Annotation analysis
Heteroplasmy and haplogroup analysis are available for request
Note: Recommended data outputs and analysis contents displayed are for reference only. For detailed information, please contact us with your customized requests.
Analysis Pipeline
Deliverables
The original sequencing data
Experimental results
Data analysis report
Details in Human Mitochondrial DNA (mtDNA) Sequencing for your writing (customization)
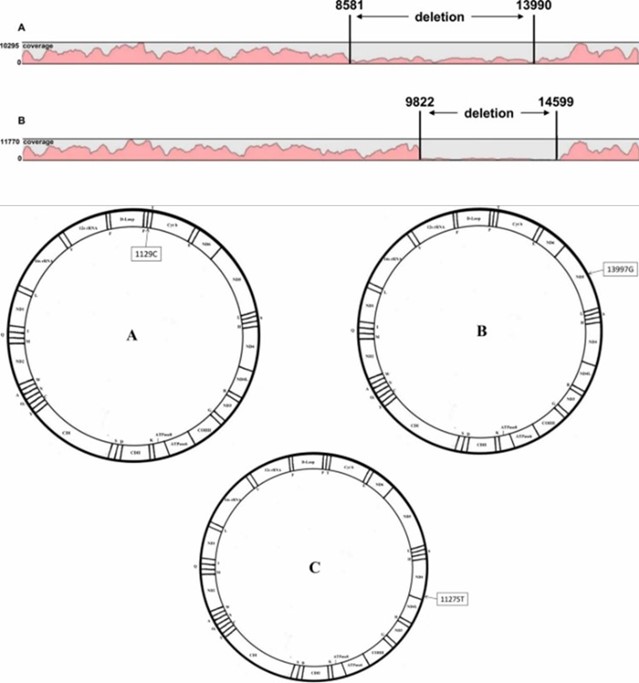
Demo Results
Mapping and coverage results of mtDNA sequencing, as well as the display of mtDNA pathological profiles. (Yao et al., 2019)
Reference:
Yao Y, Nishimura M, Murayama K, et al. A simple method for sequencing the whole human mitochondrial genome directly from samples and its application to genetic testing. Scientific Reports, 2019, 9(1): 17411.
Human Mitochondrial DNA (mtDNA) SeqFAQs
1. Does whole genome sequencing include mitochondrial DNA?
Whole genome sequencing (WGS) inherently encompasses mitochondrial DNA within its scope. This comprehensive sequencing technique involves the extraction and sequencing of both nuclear and mitochondrial DNA. Due to the abundance of mtDNA copies within cells, whole genome sequencing ensures comprehensive coverage of the mitochondrial genome, enabling concurrent analysis with the nuclear genome.
2. What are the methods for sequencing human mitochondrial DNA?
Various methodologies are employed for sequencing human mitochondrial DNA (mtDNA). Common approaches encompass Sanger sequencing, next-generation sequencing platforms (e.g., Illumina sequencing), and long-read sequencing technologies (e.g., Oxford Nanopore Technologies and Pacific Biosciences). Each methodology offers distinct advantages, drawbacks, and applicability, enabling researchers to tailor their selection based on specific research objectives and resource considerations.
Human Mitochondrial DNA (mtDNA) Seq Case Studies
A method for multiplexed full-length single-molecule sequencing of the human mitochondrial genome
Journal: Nature Communications
Impact factor: 16.7
Published: 06 October 2022
Background
Mitochondria contain circular genomes with vestigial functions. Heteroplasmy can significantly impact health, requiring accurate variant detection for diagnosis and treatment. Traditional sequencing methods have limitations, but long-read technologies like ONT and PacBio offer improved accuracy. Using Cas9 for targeted enrichment with ONT's Q20+ chemistry enhances read precision and lowers costs, making it suitable for low-quality samples and extending its use in mitochondrial research and clinical applications.
Methods
Sample Preparation:
Human cell lines
15 clinical samples
DNA quality control
Sequencing:
Long-range PCR
Circular mtDNA pre-enrichment
Library preparation
Long-read nanopore sequencing
Short-read sequencing
Data Analysis:
Pre-processing raw data
Alignment
Variant calling
Annotation of mtDNA variants
Circos plots
Results
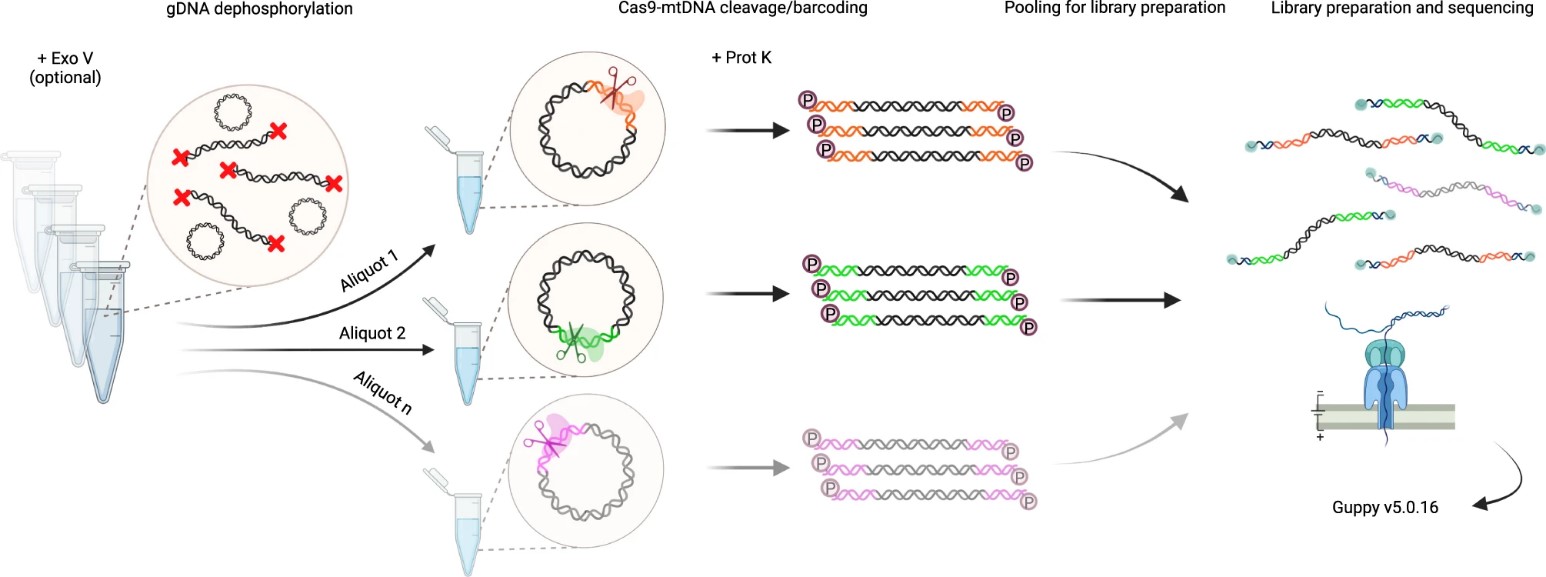
The authors refined the Cas9-mtDNA-enrichment protocol to enable highly selective single-molecule sequencing of mtDNA from human gDNA samples, facilitating multiplexing without additional barcoding steps. This method involves Exonuclease V digestion for mtDNA enrichment, dephosphorylation of gDNA ends, and sequence-specific cleavage by dual-RNA-guided Cas9 endonuclease. This approach generates a barcode at the Cas9 cut site, facilitating library preparation and sequencing. After Cas9 cleavage, samples are treated with Proteinase K to remove bound Cas9 protein, followed by ligation of ONT sequencing adaptors to phosphorylated cleavage sites.
Fig. 1 Cas9-mtDNA-enrichment, barcoding, pooling and demultiplexing approach for long-read sequencing.
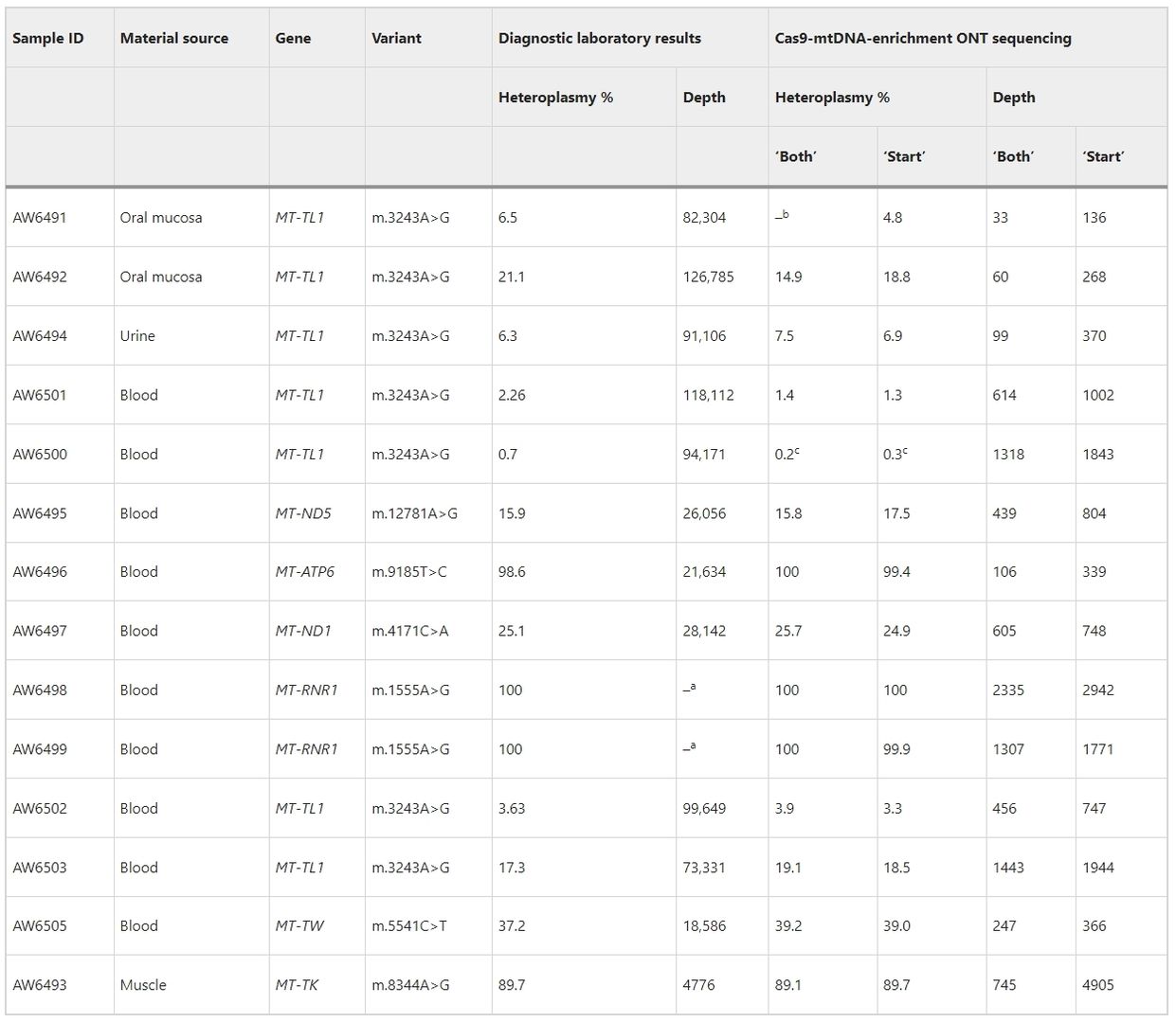
Cas9-mtDNA-enrichment analysis accurately detects heteroplasmy in pathogenic SNVs across 14 clinical samples. The method, without Exonuclease V treatment, achieves variable mtDNA genome coverage (×33 to ×2335), correlating with sample DNA degradation levels. Including both full-length reads and reads starting at specific Cas9 cut-site barcodes boosts coverage and confidence in variant calling. All reported heteroplasmies are confirmed, with frequencies ranging from <0.2% to 100%, and no large deletions observed. Additionally, non-pathogenic polymorphic sites and variants of unknown significance are identified, underscoring the importance of assessing variant frequency within the general population and specific haplogroups.
Table 1. Pathogenic SNV identification and heteroplasmy determination in the confirmed clinical samples with mtDNA alterations
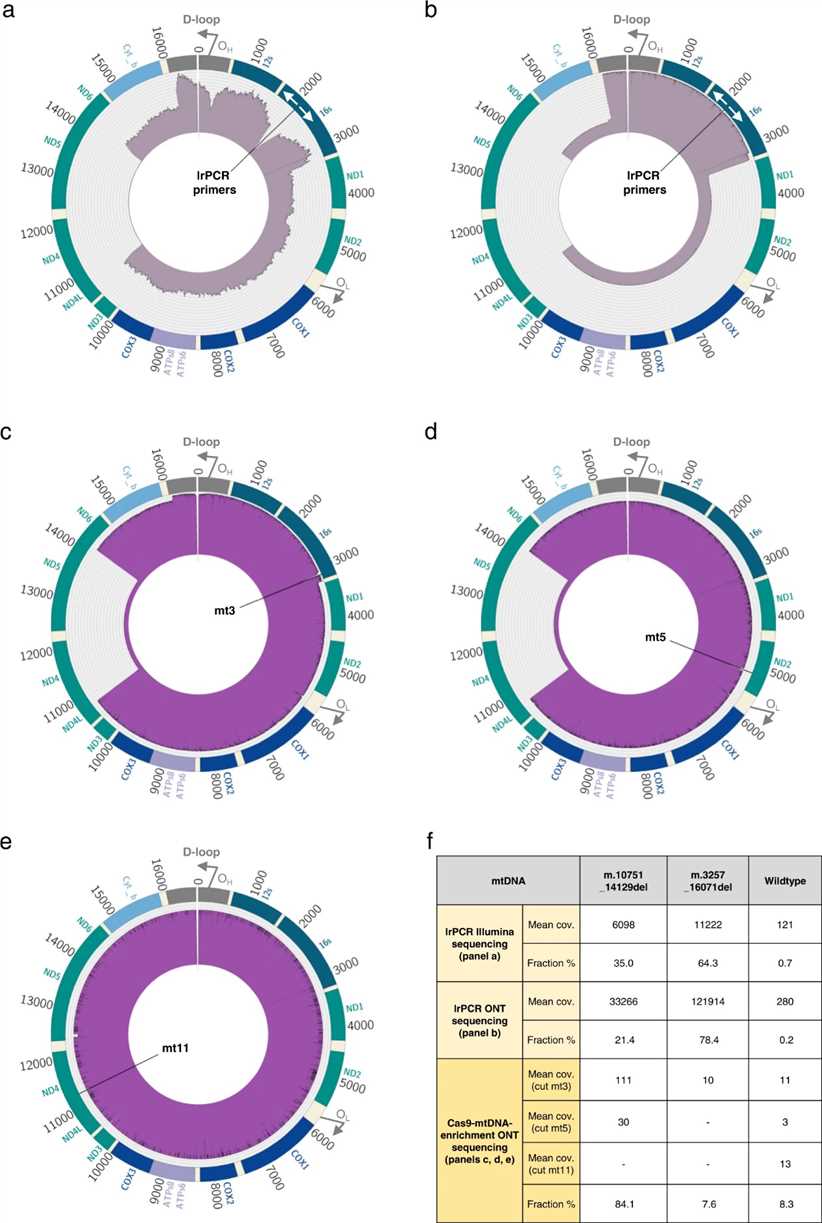
Cas9-mtDNA-enrichment analysis successfully identifies multiple mtDNA deletions in a complex clinical case. Using three gRNAs spanning the mitochondrial genome, the method detects two large deletions: m.3264_16070del (7.6% heteroplasmy) and m.10751_14129del (84.1% heteroplasmy), alongside wild-type mtDNA (8.3% frequency). Comparison with lrPCR and Illumina sequencing confirms the benefit of native mtDNA sequencing, overcoming biases and providing more accurate estimates of mtDNA populations.
Fig. 2 Multiple mtDNA deletions in a clinical sample.
Conclusion
This method uses Cas9 nuclease and nanopore sequencing to efficiently target and sequence the full-length human mitochondrial genome. It achieves high coverage, accurately detecting SNVs and multiple deletions in clinical samples. The authors' custom analysis pipeline enables precise variant calling and facilitates comprehensive mtDNA analysis, benefiting molecular diagnosis and population studies. Additionally, it has potential applications in investigating somatic mutations and DNA modifications, extending to animal species for conservation genetics and phylogenetics research.
Reference:
Keraite I, Becker P, Canevazzi D, et al. A method for multiplexed full-length single-molecule sequencing of the human mitochondrial genome. Nature Communications, 2022, 13(1): 5902.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
https://doi.org/10.1038/s41419-024-06769-5
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
https://doi.org/10.3390/ijms21249571
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
https://doi.org/10.1111/nph.14586
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
https://doi.org/10.1111/1751-7915.13411
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
https://doi.org/10.3390/v16071000
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
https://doi.org/10.3390/v12040403
See more articles published by our clients.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India