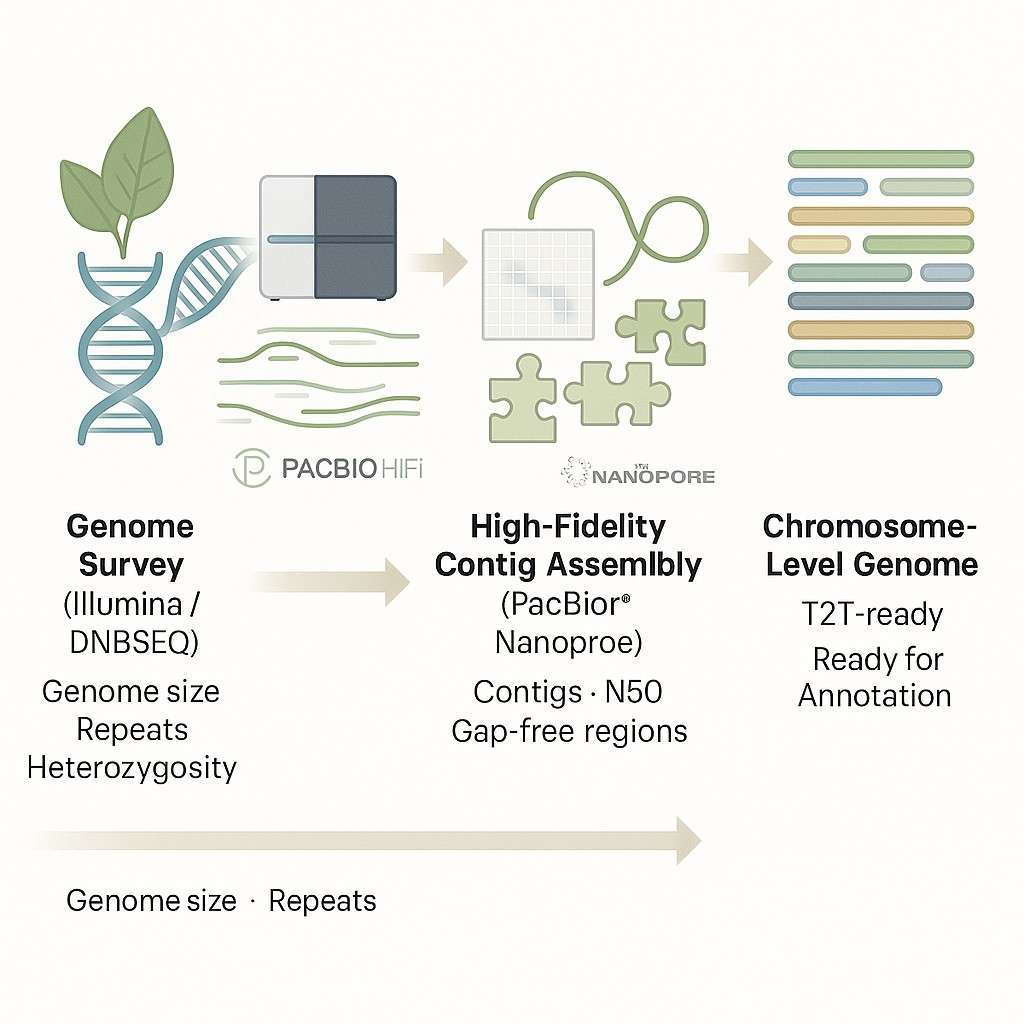
Plant/Animal Whole Genome de novo Sequencing
Home
> Plant/Animal Whole Genome de novo Sequencing
High-Resolution Genome Assembly for Plants and Animals — No Reference Required
N2Jenomics Pvt. Ltd offers end-to-end Plant and Animal Whole Genome de novo Sequencing services to decode complex genomes without relying on existing references. Our platform combines Illumina, PacBio HiFi, Oxford Nanopore, and Hi-C technologies to deliver chromosome-level assemblies with exceptional continuity and accuracy. Whether you're studying polyploid crops, wild species, or model organisms, our team provides customized strategies, expert analysis, and publication-ready data.
What Is Plant and Animal Whole Genome de novo Sequencing?
Plant and animal whole genome de novo sequencing refers to assembling a complete genome without relying on any existing reference sequence. This approach is essential when working with species that lack a reference genome, have poorly assembled genomes, or exhibit complex genomic features like high heterozygosity, polyploidy, or extensive repetitive regions.
Instead of aligning sequencing reads to a known genome, de novo assembly reconstructs the genome from scratch—like solving a massive jigsaw puzzle using only the sequence fragments generated by high-throughput sequencing platforms. The result is a high-resolution genetic blueprint that can be used for functional annotation, comparative analysis, molecular breeding, and evolutionary studies.
At N2Jenomics Pvt. Ltd , we provide end-to-end de novo sequencing services for a wide range of plant and animal species. By integrating Illumina short reads, PacBio HiFi long reads, Nanopore ultra-long reads, and Hi-C chromatin interaction data, we deliver chromosome-level genome assemblies ready for downstream research and publication.
When Should You Use De Novo Genome Sequencing?
De novo genome sequencing is the preferred strategy when no high-quality reference genome exists—or when existing references cannot meet your research objectives. Here are the most common use cases:
✅ No Reference Genome Available
For newly discovered or under-studied species, de novo sequencing enables researchers to generate a complete reference from scratch.
✅ Incomplete or Fragmented Reference
Many publicly available genomes are outdated, poorly assembled, or fragmented at the scaffold level. De novo assembly delivers chromosome-level continuity for high-resolution research.
Plant and animal genomes often contain high levels of duplication, structural variation, or repetitive elements. De novo approaches using long-read sequencing and Hi-C mapping overcome these challenges.
✅ Pan-Genome Construction
When a single reference genome cannot capture the genetic diversity of a species, building a pan-genome via de novo assembly of multiple individuals reveals population-specific variation.
✅ Trait Discovery and Molecular Breeding
High-quality assemblies provide the foundation for GWAS, QTL mapping, and genome editing—especially in agricultural, aquaculture, and livestock research.
Pro Tip:
De novo sequencing is not only for novel species. It is often the best way to upgrade a low-contiguity genome to publication-ready quality, especially when combined with HiFi and Hi-C data.
Technology Strategy Overview: Platform Comparison for Genome Assembly
Platform
Role in Assembly
Typical Coverage
Strengths
Recommended For
Illumina / DNBSEQ™
Genome survey, error correction
30–50×
High accuracy, low cost, essential for k-mer profiling
Initial genome complexity analysis
PacBio HiFi
Contig-level de novo assembly
30–60×
Ultra-high accuracy (Q20+), excellent for repeat-rich or polyploid genomes
Plant/animal genomes with high heterozygosity
Oxford Nanopore (ONT)
Gap closure, ultra-long read assembly
50–100×
Ultra-long reads (>100 kb), ideal for telomere-to-telomere (T2T) assemblies
Genomes requiring complete or near-complete continuity
Recommended Sequencing Strategy for Plant and Animal De Novo Genomes
For most plant and animal de novo genome projects, we recommend the following integrated strategy. This four-platform combination delivers chromosome-level assemblies with high contiguity and gene annotation support in a single workflow.
Step
Platform
Coverage
Purpose
1. Genome Survey
Illumina WGS
50×
K-mer analysis for genome size estimation, heterozygosity rate, and repeat content — guides downstream strategy
2. Contig Assembly
PacBio HiFi WGS
30×
High-accuracy (Q20+) long reads (15–20 kb) for primary contig backbone, spanning repeats and resolving haplotypes
3. Chromosome Anchoring
Hi-C Sequencing
100×
Chromatin conformation capture for ordering contigs into chromosome-scale pseudomolecules; typical anchoring rate >95%
4. Gene Annotation Support
RNA-seq
—
Transcriptome evidence for gene structure prediction and functional annotation validation
About Hi-C Sequencing: Hi-C (High-throughput Chromosome Conformation Capture) captures long-range chromatin interactions by crosslinking DNA in situ, digesting, and re-ligating spatially proximal fragments. The resulting chromatin interaction data allows contigs assembled from long reads to be ordered, oriented, and anchored into chromosome-scale pseudomolecules. This step is critical for achieving publication-grade chromosome-level assemblies, especially for genomes with large repetitive regions where assembly algorithms alone cannot resolve chromosomal architecture.
PacBio Revio sequencing system. Image courtesy of PacBio.
Assembly contiguity comparison: the recommended multi-platform strategy delivers chromosome-level contig N50 compared to single-platform approaches.
Hybrid Strategy Insight:
Most successful assemblies combine short reads + long reads + Hi-C. We tailor the platform mix based on genome size, ploidy, and your research goals.
De Novo Genome Sequencing Service Workflow: From Sample to Chromosome-Scale Assembly
Sample Quality Control
Integrity assessment via PFGE or Femto Pulse
Purity checks (OD ratios, Qubit, and RNA contamination removal)
Genome Survey (Illumina)
Short-read sequencing (~100X coverage)
K-mer analysis for genome size, repeat content, heterozygosity
Guides downstream long-read and Hi-C strategy
Long-Read Sequencing (PacBio HiFi or Oxford Nanopore)
High-contiguity de novo assembly of primary contigs
Platforms selected based on target genome properties
30–100X coverage depending on platform
Scaffolding and Chromosome Anchoring (Hi-C Sequencing)
Captures long-range chromatin interactions
Anchors contigs to pseudochromosomes
Enables chromosome-scale genome assembly
Polishing and Error Correction
Short-read polishing for SNP/indel correction
Gap filling and repeat resolution
BUSCO and alignment-based quality checks
Genome Annotation (Optional Add-On)
Gene structure prediction (ab initio and evidence-based)
Repeat region masking
Functional annotation (GO, KEGG, Pfam)
Bioinformatics Analysis
Our genome informatics pipeline integrates high-throughput assembly, annotation, and comparative analysis—customized for both plant and animal species. Whether you're working with a diploid, polyploid, or highly repetitive genome, we provide scalable and accurate solutions to decode complexity.
Workflow
Sample Requirements & Quality Standards
Sample Type
Required Amount
Purity Criteria
Special Notes
Fresh or frozen animal tissue
≥ 1.5 μg gDNA (≥50 kb average length)
OD260/280: 1.8–2.0; OD260/230: ≥2.0
Avoid blood-contaminated samples; no freeze-thaw cycles
For insects, remove chitin exoskeleton before extraction
Hi-C crosslinked tissue
≥ 1 g fresh tissue or ~5 million cells
OD not applicable (crosslinked)
Crosslinking and fixation must follow our Hi-C prep protocol
General QC Criteria:
High molecular weight DNA: >50 kb preferred for long-read platforms (PacBio HiFi, Oxford Nanopore)
No RNA, protein, or secondary metabolite contamination
Concentration: ≥50 ng/μL (Qubit); Integrity: Confirmed by pulsed-field gel or Femto Pulse
Need help with DNA extraction?
N2Jenomics Pvt. Ltd provides end-to-end extraction services tailored to plant and animal genomes, using magnetic bead purification to minimize shearing and contaminants. Contact us to learn more.
Deliverables
N2Jenomics Pvt. Ltd provides comprehensive and well-organized deliverables for every plant or animal whole genome de novo sequencing project. Our data packages are tailored for seamless downstream analysis and publication readiness.
✅ Standard Deliverables
File Type / Content
Description
Raw Sequencing Data
FASTQ files from PacBio HiFi, Nanopore, Illumina, and/or Hi-C platforms
Assembly Results
Genome contigs and scaffolds in FASTA format
Assembly Metrics Report
Summary of genome size, N50, GC content, completeness (BUSCO, etc.)
Contact matrices and assembly scaffolding plots (if Hi-C is included)
Circos & Synteny Plots
Visual summaries of genome architecture and comparative analysis
Bioinformatics Summary Report
Detailed methods, software versions, and pipeline descriptions
✅ Optional Add-ons (Project Upgrades)
For projects requiring advanced data analysis or tailored outputs,N2Jenomics Pvt. Ltd offers the following upgrade options:
Upgrade Option
Description
Chromosome-Level Assembly
Achieved via Hi-C or BioNano scaffolding, delivering chromosome-scale pseudomolecules
Functional Genome Annotation
Includes gene prediction, GO/KEGG enrichment, repeat elements, and TE annotations
Comparative Genomics Package
Includes whole-genome synteny, ortholog clustering, and evolutionary distance estimation
Pan-Genome Construction
Multi-sample assembly integration, structural variant detection, and shared/unique gene sets
Epigenome Integration
Add-on for methylation or histone modification maps (requires compatible sample prep)
GWAS-Ready Data Formatting
Includes SNP/INDEL calling, VCF formatting, and population structure files for GWAS pipelines
Featured Project Snapshot
Species Type
Genome Size
Contig Count
Contig N50
Hi-C Anchoring Rate
Plant A
1.02 Gb
626
7.15 Mb
95.4%
Plant B
793.46 Mb
347
34.19 Mb
96.1%
Aquatic Animal A
979.98 Mb
513
5.36 Mb
97.89%
Aquatic Animal B
827.62 Mb
170
9.88 Mb
99.51%
Mammal
3.3 Gb
2,658
79.41 Mb
98.58%
Insect
979.98 Mb
513
5.37 Mb
97.89%
These high-contiguity genomes demonstrate N2Jenomics Pvt. Ltd ' robust assembly pipeline across diverse species—from complex plant genomes to chromosome-level assemblies in mammals and aquatic organisms.
Demo Results
Below are representative data types generated during a typical plant or animal de novo genome sequencing project using the recommended Illumina + PacBio HiFi + Hi-C strategy. Results will vary by species and project scope.
Figure 1: K-mer Distribution (Genome Survey) K-mer frequency plot from Illumina short-read data (~50×), used to estimate genome size, heterozygosity rate, and repeat content prior to long-read sequencing.
Figure 2: Hi-C Chromatin Interaction Heatmap Genome-wide Hi-C contact map (100×) used to order and orient contigs into chromosome-scale pseudomolecules. Strong diagonal signal confirms correct scaffolding.
Figure 3: BUSCO Completeness Assessment Percentage of complete, fragmented, duplicated, and missing BUSCO genes. Reference-quality assemblies typically achieve >95% complete BUSCO scores.
Figure 4: Assembly Contiguity Comparison Contig N50 comparison across different sequencing strategies. The hybrid approach combining PacBio HiFi + Hi-C consistently produces the highest contiguity for plant and animal genomes.
Reference
Hotaling, et al. Highly accurate long reads are crucial for realizing the potential of biodiversity genomics. BMC Genomics. 2023. https://doi.org/10.1186/s12864-023-09193-9
Plant/Animal Whole Genome de novo Seq FAQs
What is Plant or Animal Whole Genome de novo Sequencing?
It's a reference-free approach to reconstructing a species' entire genome from scratch. This method is essential for species lacking a reliable reference genome or those with complex structural variations.
When should I choose de novo genome sequencing over resequencing?
Answer:
Choose de novo sequencing when:
No high-quality reference genome exists.
Your species has significant genomic diversity or complexity.
You aim to build a pan-genome or improve current reference quality.
What sequencing platforms are used in your service?
Plant/Animal Whole Genome de novo Seq Case Studies
Case Study: Deciphering m6A Methylation Mechanisms in Arabidopsis Using Whole Genome de novo Sequencing
Journal: New Phytologist Impact Factor: 8.3 Published: 2017 DOI: 10.1111/nph.14586
Background
As a model organism for plant genetics, Arabidopsis thaliana has been instrumental in uncovering epigenetic regulatory mechanisms. Among these, N6-methyladenosine (m6A) modification of mRNA plays a pivotal role in plant growth, development, and stress responses. However, the molecular components driving this modification—and their functional conservation in higher plants—remain incompletely understood.
This study aimed to identify the genetic factors essential for m6A RNA methylation inArabidopsis by integrating whole genome de novo sequencing with targeted functional genomics. A central focus was placed on understanding the role of HAKAI, a conserved E3 ubiquitin ligase, within the methylation machinery.
Materials & Methods
Genome Analysis and Mutant Screening:
Arabidopsis thaliana mutants with suspected m6A methylation defects were selected from T-DNA insertion libraries.
Genomic DNA was extracted and sequenced de novo using Illumina short-read and ONT long-read platforms, achieving high contiguity and coverage.
Gene disruption events were mapped and validated.
m6A Profiling:
Total RNA was isolated from mutant and wild-type lines.
m6A quantification was performed using LC-MS/MS and immunoprecipitation-based m6A-seq.
Functional Validation:
Complementation assays were used to verify gene function.
RNA-seq was applied to assess transcriptomic consequences of HAKAI loss.
Results
The whole genome de novo sequencing enabled accurate identification of T-DNA insertions disrupting HAKAI, a gene encoding a RING-domain E3 ubiquitin ligase. Functional loss of HAKAI significantly reduced global m6A methylation levels, comparable to mutants of known m6A writers such as MTA and FIP37.
Key Findings:
Loss of HAKAI led to defects in apical dominance, flowering time, and embryo viability, phenocopying other core m6A component mutants.
Transcriptome analysis revealed dysregulation in key developmental and hormone signaling pathways.
Complementation of the HAKAI gene restored both m6A methylation levels and normal development.
Conclusion
This study demonstrated that HAKAI is a critical component of the m6A methylation complex in plants, acting alongside canonical methyltransferases. The use of whole genome de novo sequencing allowed precise mapping of gene disruptions and was essential for validating functional hypotheses in genetically complex backgrounds.
The case highlights how plant whole genome de novo sequencing, paired with epitranscriptomic and transcriptomic tools, can unravel conserved regulatory mechanisms. N2Jenomics Pvt. Ltd supports similar studies by offering integrated genome assembly, methylation analysis, and functional genomics pipelines for plant epigenetics and beyond.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Combinations of Bacteriophage Are Efficacious against Multidrug-ResistantPseudomonas aeruginosaand Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
https://doi.org/10.3390/v16071000
Genome sequence, antibiotic resistance genes, and plasmids in a monophasic variant ofSalmonella typhimuriumisolated from retail pork
Journal: Microbiology Resource Announcements
Year: 2024
https://doi.org/10.1128/mra.00754-23
Genes ofSalmonella entericaSerovar Enteritidis Involved in Biofilm Formation
Journal: Applied Microbiology
Year: 2024
https://doi.org/10.3390/applmicrobiol4020053
Complete genome sequence of the probioticBifidobacterium adolescentisstrain iVS-1
Journal: Microbiology Resource Announcements
Year: 2023
https://doi.org/10.1128/MRA.00541-23
See more articles published by our clients.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India