CiFi Sequencing Service
Home
> CiFi Sequencing Service
CiFi Sequencing Service — Long-Read Chromosome Conformation Capture with PacBio HiFi
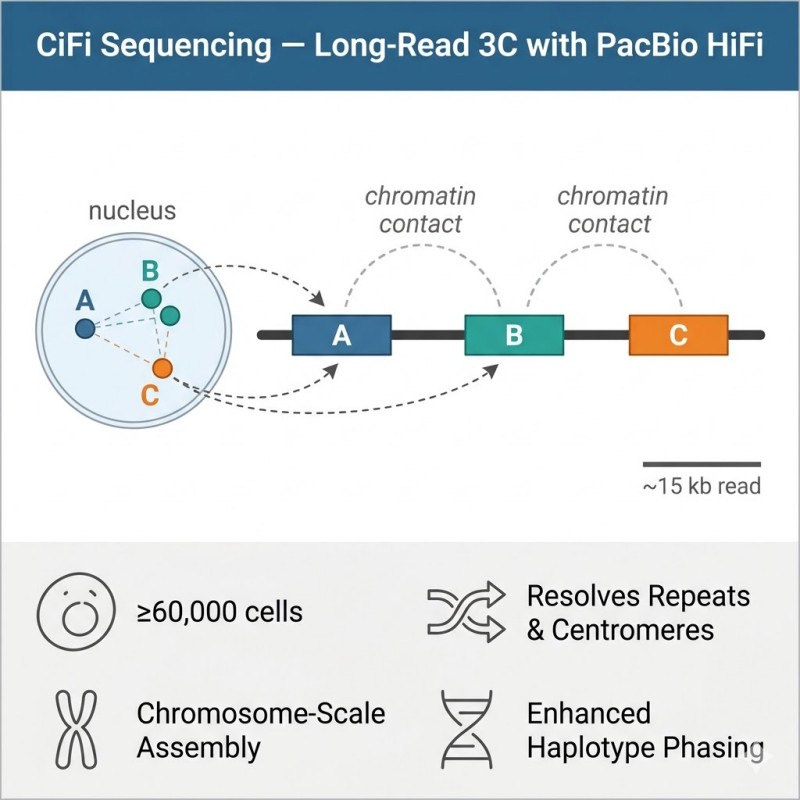
N2Jenomics Lab Pvt. Ltd. delivers CiFi sequencing: a next-generation chromatin conformation capture method that couples 3C with PacBio HiFi long-read sequencing. Starting from as little as 60,000 cells, CiFi generates multi-kilobase concatemer reads carrying multiple interacting segments, enabling accurate TAD analysis, haplotype phasing, and chromosome-scale genome scaffolding across regions where standard Hi-C fails.
Ultra-low input: ≥60,000 cells (sub-microgram DNA) — compatible with single small insects
CiFi (Chromosome conformation capture with HiFi sequencing) is a method that integrates chromosome conformation capture (3C) chemistry with PacBio HiFi long-read sequencing. Published in Nature Communications in December 2025 (McGinty et al.), CiFi was developed to overcome the two core limitations of conventional Hi-C sequencing: high cell-input requirements and poor mappability in repetitive genomic regions.
Standard short-read Hi-C requires millions of cells and produces paired 150 bp reads. When those reads fall within centromeres, segmental duplications, or other repeat-rich regions, they cannot be uniquely mapped — leaving the most biologically significant parts of the genome invisible to contact analysis. CiFi addresses both problems simultaneously. By applying whole-genome amplification to 3C libraries and sequencing the resulting long concatemers on PacBio HiFi, CiFi generates multi-kilobase reads (5–25 kb) that each carry multiple interacting genomic segments (350 bp to 2 kb per segment). These longer, multi-contact reads map uniquely across regions that defeat short reads, and the library preparation is sensitive enough to work from sub-microgram DNA inputs — as few as 60,000 cells.
The resulting data are largely concordant with conventional Hi-C for standard interaction detection, but unlock a series of additional capabilities: TAD boundary calling across centromeres, enhanced haplotype phasing, and chromosome-scale genome assembly scaffolding — all from a single sequencing run, optionally combined with HiFi whole-genome sequencing on the same instrument.
CiFi vs. Standard Hi-C: Where Long Reads Win
Both methods capture proximity-ligation chromatin contacts genome-wide. CiFi adds long-read accuracy, multi-contact information, and low-input sensitivity that short-read Hi-C cannot deliver.
Feature
Standard Hi-C (Short-Read)
CiFi (PacBio HiFi Long-Read)
Minimum cell input
Typically ≥1 million cells
≥60,000 cells (sub-microgram DNA)
Read length
150 bp paired-end
5–25 kb multi-contact HiFi reads
Contacts per read
2 segments per read pair
Multiple segments per read (multi-contact)
Repetitive region mappability
Poor — centromeres, SDs, low-complexity regions missed
Improved — longer segments span repeats uniquely
Haplotype phasing
Limited — short reads rarely span heterozygous SNPs
Substantially enhanced — HiFi accuracy across phased blocks
TAD analysis in complex regions
Fails at centromeres and segmental duplications
Resolves TADs across disease-associated genomic hotspots
Genome assembly scaffolding
Compatible with Hi-C scaffolders
Competitive or fewer contacts needed vs. standard Hi-C
Co-sequencing with WGS
Requires separate library and sequencing run
Single SMRT Cell can produce WGS + CiFi library simultaneously
Small organism / single individual
Not feasible without pooling
Single small insect validated (Anopheles mosquito, fruit fly)
Core Technical Capabilities
CiFi combines PacBio HiFi accuracy with 3C multi-contact library chemistry to deliver four integrated capabilities from a single experiment.
Ultra-Low Input Chromatin Profiling
Starting material: ≥60,000 cells or sub-microgram genomic DNA
Whole-genome amplification (WGA) of 3C libraries preserves contact information while amplifying limited input
Validated on single insects (Anopheles coluzzii mosquito; Ceratitis capitata Mediterranean fruit fly)
Opens chromatin 3D profiling to previously inaccessible sample types: rare cell populations, precious biopsies, individual small organisms
Repetitive Region Resolution
Segments of 350 bp to 2 kb per concatemer enable unique mapping in centromeres and segmental duplications
Gains in TAD/domain boundary calling across human disease-associated genomic hotspots
Improved coverage uniformity across satellite repeat arrays, tandem duplications, and low-complexity loci
Pairwise interactions concordant with conventional Hi-C for euchromatic regions, with additional resolution in heterochromatin
Haplotype-Resolved Phasing
HiFi base accuracy (>99%) across phased genomic segments enables confident heterozygous SNP calling within contact reads
Multi-contact reads provide long-range phase blocks substantially improved over short-read Hi-C
Phase block extension across complex structural variants and repeat boundaries
CiFi contacts scaffold HiFi contig assemblies to chromosome scale — same data pipeline as Hi-C scaffolding, improved by longer reads
Demonstrated: chromosome-scale phased diploid assembly of a single Mediterranean fruit fly from one SMRT Cell
Compatible or lower contact requirements vs. traditional Hi-C for scaffolding equivalent genome sizes
Enables single-individual genome projects for organisms with limited material
Service Workflow
From sample submission to publication-ready chromatin interaction data and genome assembly outputs.
Step 1 — Consultation & Sample QC: We review your genome size, organism, available cell number, and project goals (3D chromatin analysis only, or combined WGS+CiFi single-run assembly). All samples undergo cell count verification and nucleic acid QC before processing. Detailed sample handling guidance is provided upon project initiation to preserve chromatin integrity during shipping.
Step 2 — 3C Library Preparation: Cells are crosslinked with formaldehyde to preserve chromatin contacts, digested with a frequent-cutting restriction enzyme (DpnII or HindIII depending on genome GC content), and proximity-ligated under dilute conditions that favor in situ junctions reflecting real spatial proximity. Long ligated fragments are purified and size-selected to maximize multi-kb concatemer yield.
Step 3 — WGA Amplification & PacBio HiFi Sequencing: Whole-genome amplification is applied to the 3C library to reach PacBio input requirements without losing contact information. PacBio-compatible SMRTbell adaptors are ligated and libraries are sequenced on PacBio Revio using circular consensus sequencing (CCS/HiFi) mode, generating per-read accuracy >99% across multi-kb concatemers. When combined with HiFi WGS, both libraries can be processed from the same individual.
Step 4 — Multi-Contact Bioinformatics Analysis: HiFi reads are processed to extract individual segments, assign V(D)J-equivalent contact pairs, and generate genome-wide interaction maps. TAD/compartment/loop calling is performed with established tools (juicer, HOMER, or equivalent). Haplotype phasing integrates CiFi multi-contact information with HiFi contig phase blocks. For assembly projects, CiFi contacts scaffold HiFi contigs to chromosome scale using 3D-DNA or YaHS.
Step 5 — Results Delivery: You receive raw HiFi FASTQ files, per-sample QC metrics, pairwise interaction matrices (.cool/.hic), chromatin domain annotation files, haplotype phasing output, and — for assembly projects — a chromosome-scale scaffolded FASTA with assembly statistics. A scientific consultation call is included to discuss results and downstream applications.
Demo Results
Genome-wide CiFi contact matrix (human GM12878 lymphoblastoid cell line) showing improved interaction coverage across centromeric and segmental duplication regions (highlighted) compared to standard short-read Hi-C. (McGinty SP et al., Nat Commun, 2025)
Chromosome-scale phased diploid assembly of a single Mediterranean fruit fly (Ceratitis capitata) produced by combining HiFi whole-genome sequencing and CiFi contacts from one individual. CiFi scaffolded HiFi contigs into chromosome-scale sequences. (McGinty SP et al., Nat Commun, 2025)
CiFi Sequencing FAQs
1. How does CiFi differ from Pore-C or standard HiFi-C methods?
CiFi, Pore-C, and HiFi-C all combine long-read sequencing with 3C chemistry, but differ in key aspects. CiFi uses PacBio HiFi sequencing with whole-genome amplification of 3C libraries, achieving >99% per-read accuracy and enabling sub-microgram input. Pore-C uses Oxford Nanopore without amplification, requiring higher input and trading base-level accuracy for longer raw reads. HiFi-C is the broader term for PacBio-based 3C approaches, of which CiFi is a specific validated protocol with published performance benchmarks in Nature Communications. N2Jenomics Lab Pvt. Ltd. implements the CiFi protocol with the WGA amplification step that enables the 60,000-cell minimum input.
2. Can CiFi be used without a reference genome?
Yes. For organisms without a reference, CiFi contacts scaffold de novo HiFi contig assemblies to chromosome scale — the CiFi data effectively serves as the phasing and scaffolding layer on top of a HiFi-based draft assembly. This is the application demonstrated with the Ceratitis capitata Mediterranean fruit fly in the original CiFi publication. When sequencing a new organism, we recommend planning a combined WGS+CiFi project from the same individual to streamline data integration.
3. What organisms has CiFi been validated on?
The original CiFi paper validated the method on human lymphoblastoid cells (GM12878), a single Anopheles coluzzii mosquito (genome-wide chromatin interaction profiling), and a single Ceratitis capitata Mediterranean fruit fly (chromosome-scale phased diploid assembly). The method is organism-agnostic and applicable to any species where cells or tissue can be crosslinked for 3C library preparation. We have applied Hi-C and related long-read methods to a broad range of insects, plants, vertebrates, and fungi; contact us to discuss your specific organism.
4. Is CiFi compatible with FFPE or archived samples?
CiFi requires intact crosslinked chromatin for 3C library preparation, which means it requires fresh or flash-frozen tissue — not FFPE. Ethanol-preserved museum specimens (insects, small organisms) have been used successfully when handled with appropriate care. If your material is FFPE, we can advise on alternative chromatin capture strategies or alternative sequencing approaches for genomic analysis from archived material.
5. Can you perform CiFi from sorted cell populations or flow cytometry-isolated cells?
Yes. FACS-sorted cell populations are compatible with CiFi as long as sorting is performed under conditions that preserve cell membrane integrity for downstream crosslinking, and the recovered cell number meets the 60,000-cell minimum. We recommend coordinating the sorting protocol with our scientific team before processing to ensure chromatin integrity is maintained throughout.
CiFi Sequencing Case Studies
Customer Publication Highlight
Unraveling Forensic Timelines Using Molecular Markers in Phormia regina Maggots
Journal: PLOS Genetics Impact Factor: 3.7 Published: 2025 Service Used: Hi-C Library Construction and Sequencing
Background
Estimating the post-mortem interval (PMI) in forensic investigations relies critically on understanding the development timeline of blow flies, particularly Phormia regina. Accurate molecular dating of developmental stages — from egg to pupal stage — requires access to high-quality genomic and transcriptomic reference data. However, generating chromosome-scale genome assemblies for insects is challenging due to the limited tissue available from individual specimens and the complexity of repetitive regions in dipteran genomes. This study employed Hi-C library construction and sequencing to provide the long-range contact data needed for chromosome-scale genome scaffolding, enabling subsequent transcriptomic profiling of developmental markers at each forensic-relevant life stage.
Materials & Methods
Sample Preparation
Phormia regina blow fly specimens across developmental stages
Fresh tissue collection for Hi-C crosslinking
RNA extraction for transcriptome profiling at each developmental timepoint
Sequencing
Hi-C library construction and sequencing (N2Jenomics Lab Pvt. Ltd. service)
High-throughput chromosome conformation capture for genome scaffolding
Short-read and/or long-read WGS for contig assembly
Transcriptomic marker profiling across forensically relevant developmental stages
PMI estimation model building from molecular timeline data
Results
Hi-C-Scaffolded Chromosome-Scale Genome Assembly
Hi-C Library Construction and Sequencing from N2Jenomics Lab Pvt. Ltd. provided the chromatin contact data needed to scaffold the P. regina genome to chromosome scale, enabling reliable gene-level annotation essential for downstream transcriptomic analyses.
The chromosome-scale assembly resolved repetitive regions characteristic of dipteran genomes, providing a high-quality reference for developmental marker identification.
Molecular Marker Identification for Forensic PMI Estimation
Using the scaffolded genome as reference, transcriptomic profiling identified stage-specific molecular markers across the blow fly developmental timeline — from egg through late larval and pupal stages.
These markers enabled construction of a PMI estimation model based on gene expression patterns, providing forensic scientists with a molecular-level alternative to morphology-based developmental staging.
Hi-C-assisted chromosome-scale genome assembly and developmental molecular marker profiling in Phormia regina — enabling molecular PMI estimation for forensic applications. (PLOS Genetics, 2025, DOI: 10.1371/journal.pgen.1011948)
Conclusion
This study demonstrates the direct utility of Hi-C Library Construction and Sequencing for chromosome-scale genome assembly in forensically and ecologically important insects. The approach mirrors the core CiFi use case: a single insect species with complex repetitive genomic regions, limited tissue availability, and a need for high-quality chromosome-scale assembly to enable downstream molecular research. CiFi extends this capability further — enabling the same genome assembly workflow from input as low as a single individual, without pooling specimens.
Reference
Unraveling forensic timelines using molecular markers in Phormia regina maggots. PLOS Genetics. 2025. https://doi.org/10.1371/journal.pgen.1011948
Related Publications
Here are some publications from our clients that used our Hi-C Library Construction and Sequencing service:
Unraveling forensic timelines using molecular markers in Phormia regina maggots
Journal: PLOS Genetics
Year: 2025
https://doi.org/10.1371/journal.pgen.1011948
See more articles published by our clients.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India