Polysome Sequencing Service
Home
> Polysome Sequencing Service
Polysome Sequencing Service: Unveiling Translational Dynamics with Precision and Depth
In the rapidly advancing field of molecular biology, understanding gene expression demands more than simply analyzing mRNA levels—it requires direct insights into the translation process. Our Polysome Sequencing Service empowers researchers to dissect translational regulation with unparalleled resolution, revealing how mRNAs engage with ribosomes under diverse biological conditions. Whether investigating disease mechanisms, RNA modifications, or hidden coding potentials in non-coding RNAs, we provide a comprehensive solution tailored for cutting-edge translational research.
Quantifying translation efficiency (TE) for thousands of transcripts simultaneously.
Identifying actively translated non-coding RNAs, such as lncRNAs and circRNAs.
Profiling translational shifts in response to cellular stress, disease states, or therapeutic interventions.
Delivering precise, multi-tiered data ready for integrative omics analysis.
In modern molecular biology, gene expression research has moved far beyond simply quantifying mRNA levels. The translation process—where ribosomes decode mRNA blueprints into proteins—accounts for over half of all gene regulatory events, exerting a profound influence on cellular behavior, protein function, and disease development. Yet traditional transcriptomic methods often fall short in revealing the true landscape of protein synthesis, creating gaps between measured mRNA abundance and actual protein production.
Polysome sequencing (Polysome-seq) bridges this critical gap. By combining polysome profiling with high-throughput sequencing, Polysome-seq offers researchers a comprehensive, quantitative snapshot of how ribosomes engage with thousands of mRNAs across diverse biological conditions. It unveils dynamic insights into translational regulation, enabling precise exploration of gene expression at the level where proteins—the ultimate effectors of cellular function—are actually produced.
How Polysome Profiling Works
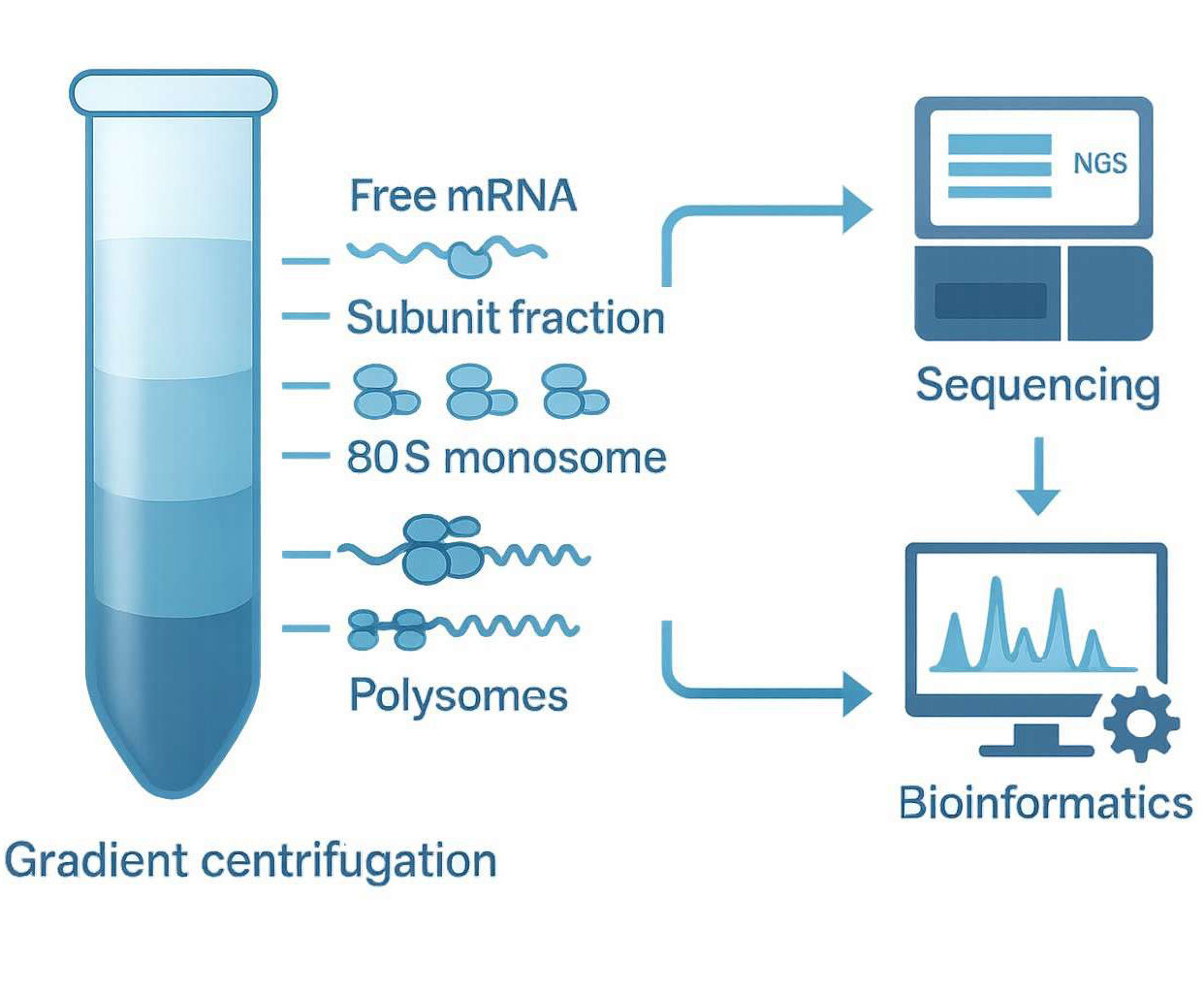
Polysome profiling is an analytical method that separates cytoplasmic RNA based on the number of ribosomes bound to each mRNA molecule. Utilizing sucrose gradient ultracentrifugation, cellular lysates are fractionated into distinct layers representing:
Free mRNA (not engaged in translation)
40S and 60S ribosomal subunits
80S monosomes (single ribosomes)
Light and heavy polysomes (multiple ribosomes translating a single mRNA)
A Comparison of Translational Omics Technologies
Translational research has expanded to include several high-resolution technologies, each offering unique insights:
Technology
Core Focus
Key Advantages
Limitations
Polysome Profiling / Polysome-seq
Measures ribosome occupancy to infer translation efficiency.
- Direct translation efficiency measurement - Retains longer RNA fragments for downstream analysis
- Larger sample input required - No ribosome positional data
Ribo-seq (Ribosome Profiling)
Maps precise ribosome positions on mRNA at codon resolution.
- Preserves entire mRNA structure - Detects alternative splicing isoforms
- Lacks ribosome positional information - Lower resolution of translation dynamics
Disome-seq
Detects ribosome collisions and translational pauses.
- Illuminates co-translational regulatory events
- Specialized, newer technique with fewer applications
TRAP-seq
Isolates ribosome-bound mRNAs in specific cell types via tagged ribosomes.
- Cell- or tissue-specific translation profiling
- Requires transgenic models - Possible interference with ribosome function
Among these technologies, Polysome-seq stands out as an ideal compromise—it preserves RNA integrity, reveals translation efficiency across the transcriptome, and enables integrative analysis alongside transcriptomics, epitranscriptomics, and proteomics.
We offer a full suite of translational profiling services, including Polysome-seq, Ribo-seq, RNC-seq, Disome-seq, and TRAP-seq, to meet diverse research needs across all areas of molecular biology.
Our Polysome Sequencing Workflow: From Sample to Insight
1. Consultation & Experimental Design
Every project starts with a detailed discussion between our scientific team and your research group to:
Define your biological questions and hypotheses.
Select appropriate experimental conditions, controls, and replicates.
Choose between single-fraction or multi-fraction polysome sequencing strategies.
Align your project with publication-quality standards.
2. Sample Preparation & Ribosome Stabilization
Cells or tissues are harvested and treated with translation elongation inhibitors (e.g., cycloheximide) to freeze ribosomes in place.
Rapid processing under RNase-free conditions preserves ribosome-mRNA complexes and prevents degradation.
3. Polysome Profiling via Sucrose Gradient Ultracentrifugation
Cytoplasmic extracts are layered onto a linear sucrose gradient (typically 10-50%).
Ultracentrifugation separates mRNA-ribosome complexes based on density, yielding fractions that distinguish.:
UV absorbance profiling precisely defines the peaks corresponding to each fraction.
4. Fraction Collection & RNA Extraction
Individual gradient fractions are collected, either:
As a single "pooled polysome fraction" (for cost-effective global profiling).
Or as multiple fractions (light vs. heavy polysomes) for deeper resolution of translational shifts.
RNA is extracted from each fraction, ensuring high integrity for downstream sequencing.
Microbial Physiology – Analyze bacterial and fungal translational regulation under environmental changes.
Non-Coding RNA Translation – Detect hidden peptides from lncRNAs, circRNAs, and other ncRNAs.
Stress Response Mechanisms – Profile global translation changes under heat shock, oxidative stress, or hypoxia.
Drug Mechanism Studies – Assess how therapeutic compounds impact translational efficiency and ribosome engagement.
Demo Results
Polysome Seq FAQs
1. What is Polysome‑Seq and how does it work?
Polysome‑Seq merges polysome profiling with RNA sequencing to assess translational status across the transcriptome—separating light and heavy ribosome‑associated fractions and quantifying their mRNA content for translation efficiency analysis.
2. How does Polysome‑Seq differ from Ribo‑seq?
Polysome‑Seq profiles the number of ribosomes on each mRNA, offering a view of global translation. In contrast, Ribo‑seq maps ribosome footprint positions at codon resolution—ideal for detecting start sites, uORFs, and paused translation.
3. Can Polysome‑Seq detect translation of non-coding RNAs?
Yes. By sequencing longer mRNA fragments from polysome fractions, Polysome‑Seq can capture actively translated lncRNAs, circRNAs, and other ncRNAs, revealing hidden peptides.
4. What sample types are compatible with Polysome‑Seq?
Common samples include cultured cells, animal or plant tissues, bacteria, fungi, and even purified ribosome complexes. Proper sample handling and ribosome stabilization are essential.
5. Is specialized equipment required?
Yes. Polysome profiling relies on ultracentrifugation, gradient fractionation, and UV detection. These steps demand technical expertise and high-quality reagents—precisely what our team provides.
6. What bioinformatics analyses are included?
Our pipeline includes data QC, genome/transcriptome alignment, transcript quantification, TE calculation, differential translation analysis, and visualizations. Integration with RNA‑seq, epitranscriptomics, or proteomics is also available for a comprehensive translational profile.
7. What are the limitations of Polysome‑Seq?
Potential challenges include large sample input requirements and moderate RNA recovery efficiency. Additionally, positional ribosome data is not provided—unlike Ribo‑seq.
8. Can Polysome‑Seq and Ribo‑seq be used together?
Absolutely. Combining both yields a holistic view: Polysome‑Seq reveals translational engagement, while Ribo‑seq offers detail on ribosome positioning and non-canonical translation events.
9. How is polysome profile data interpreted?
Profiles display ribosome distribution across mRNA via UV absorbance peaks. The ratio of polysomes to monosomes indicates translational activity. Subsequent sequencing enables quantitative translation efficiency analysis.
10. What quality control measures are in place?
We implement rigorous checks at each stage—UV profile reproducibility, RNA integrity (RIN score), library quality metrics, and bioinformatics quality control. These ensure reliable, high-impact results.
Polysome Seq Case Studies
Title: METTL5 stabilizes c-Myc by facilitating USP5 translation to reprogram glucose metabolism and promote hepatocellular carcinoma progression
Source: Xia et al., Cancer Communications, 2023 Impact Factor: 20.1
Study Background
METTL5 is an 18S rRNA methyltransferase.
Scientists suspected METTL5 might promote cancer by affecting how certain mRNAs are translated into proteins.
Focus was on c-Myc, a critical oncogene, and USP5, a deubiquitinase that stabilizes c-Myc.
Key Research Questions
Does METTL5 affect c-Myc protein levels through translational regulation?
How does this influence cancer cell metabolism?
Methods Used
✅ Polysome Profiling + RNA-Seq
Separated mRNAs into fractions based on ribosome loading.
Sequenced polysome-bound mRNAs to assess translation efficiency.
✅ qRT-PCR and Western Blot
Verified changes in RNA levels and protein levels.
✅ ChIP Assay & Metabolomics
Explored regulatory mechanisms and metabolic shifts.
✅ Patient-Derived Xenografts (PDX)
Validated findings in animal tumor models.
Major Findings
METTL5 upregulates translation of USP5 mRNA, resulting in higher USP5 protein levels.
USP5 stabilizes c-Myc protein by reducing its degradation.
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India