Structural Variant and Haplotype Analysis Solution
Home
> Structural Variant and Haplotype Analysis Solution
Structural Variant and Haplotype Analysis Solution
Structural variants and haplotypes often explain genomic patterns that SNP-only analysis cannot fully resolve. N2Jenomics Lab Pvt. Ltd. provides a Structural Variant and Haplotype Analysis Solution that connects sequencing strategy, SV detection, haplotype phasing, annotation, visualization, and custom bioinformatics into one research-focused workflow.
We help you move from raw genome variation data to results your team can review, discuss, and use for the next research decision. This solution is especially useful for population studies, trait mapping, non-model organism research, strain comparison, and complex genome analysis.
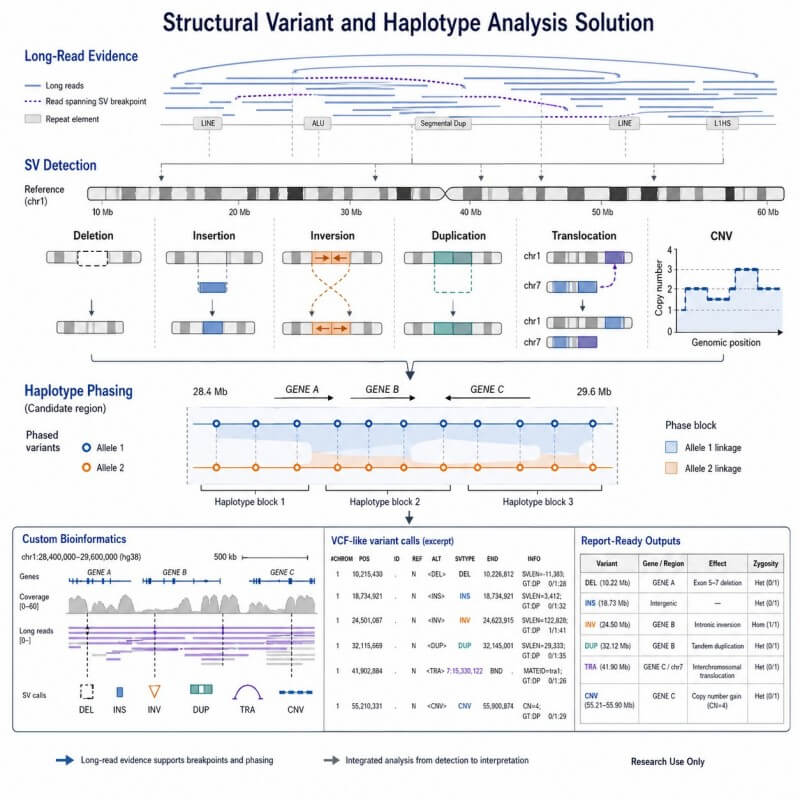
Detect deletions, insertions, inversions, duplications, translocations, and CNVs
Resolve phased variant context across candidate regions
Integrate SV and haplotype results with GWAS, QTL-seq, BSA, or population analysis
When SNPs Are Not Enough: Why Structural Variants and Haplotypes Matter
SNPs and small indels are useful, but they are only one layer of genome variation. Many research questions depend on larger genome changes, such as copy number shifts, large insertions, inversions, translocations, repeat-associated variants, or allele-specific variant combinations.
A structural variant and haplotype analysis project looks beyond isolated base changes. It asks a more practical question: what genome architecture is linked to the biological difference you are studying?
Structural variants can affect gene dosage, coding sequences, regulatory regions, genome organization, and candidate intervals. Haplotype analysis adds another layer by showing which variants occur together on the same allele or genomic background. That context can be important when a research signal depends on inherited blocks, parental alleles, population structure, strain differences, or cultivar-level variation.
Long-read sequencing is often valuable in this area because long reads can span repetitive regions, complex rearrangements, and linked variants that short reads may not resolve well. Recent studies have shown that long-read sequencing can improve structural variant discovery and haplotype-aware analysis in difficult genomic regions.
For many teams, the main question is not simply whether SVs are present. The more useful question is whether SVs and haplotypes can be connected to candidate genes, candidate regions, group differences, or downstream interpretation. That is where a planned solution matters.
What This Solution Helps You Resolve
Our Structural Variant and Haplotype Analysis Solution is designed for projects where standard variant analysis leaves important questions open.
Organize phased evidence around the research question
Population, strain, cultivar, or germplasm comparison
In Population Genetics, breeding research, and strain-level studies, the same gene region may carry different structural forms across groups.
Compare SV patterns across groups
Summarize haplotype structures by population or line
Support germplasm and diversity studies
Non-model organism and complex genome analysis
Many plants, animals, microbes, and environmental organisms have repetitive regions, variable reference quality, high heterozygosity, polyploidy, or incomplete genome resources.
Review genome size and reference status
Assess sample quality before platform selection
Adapt analysis to genome complexity
Integration with downstream analysis
SV and haplotype results are most useful when they connect to the rest of the study.
Our Service Capabilities for SV and Haplotype Projects
We do not treat SV and haplotype analysis as a single standard pipeline. A useful project plan depends on your sample, species, genome structure, existing data, and biological question.
Sequencing strategy design
We review whether your project is better suited for short-read sequencing, long-read sequencing, or a hybrid strategy. Projects focused on large SVs, repeats, haplotypes, complex loci, or non-model genomes often benefit from long-read evidence.
Haplotype phasing and haplotype-aware interpretation
Haplotype phasing helps organize variants by allele or genomic background. This can help your team understand whether variants are linked, how they differ between groups, and whether a candidate region contains phased variant patterns that matter for interpretation.
We can prepare outputs that help your team review and communicate the results, including SV summary tables, phased variant files, annotation tables, genome browser tracks, summary figures, and project reports.
Technology Strategy: Long-Read, Short-Read, or Hybrid?
The best strategy depends on what you need to resolve. No single platform or analysis method is best for every SV and haplotype project. A 2024 Nature Communications benchmark comparing alignment-based and assembly-based long-read SV detection methods found clear tradeoffs. Assembly-based methods performed well for large SVs, especially insertions, while alignment-based methods showed advantages for genotyping accuracy at lower coverage and for some complex SV classes. The study also emphasized that there is no universally superior tool across all scenarios.
Strategy
Best fit
SV detection value
Haplotype value
Sample sensitivity
Bioinformatics needs
Practical notes
Short-read WGS
SNP/Indel discovery, broad resequencing, existing cohort data
Limited for large or complex SVs; useful for small variants and supporting evidence
Limited phasing unless supported by additional data
Generally more tolerant of fragmented DNA than long-read workflows
Standard variant calling, filtering, annotation
Useful when cohort-level small variant data are needed
PacBio HiFi long-read sequencing
Accurate long-read variant discovery, complex regions, haplotype-aware analysis
Strong for insertions, deletions, repeat-associated variants, and complex regions
Strong when read length and accuracy support phasing
Best when a reference-level or allele-resolved genome foundation is needed
End-to-End Workflow with QC Checkpoints
From project intake to report-ready SV and haplotype results
We start by reviewing your species, sample type, number of samples, reference genome status, research goal, target variant types, and downstream analysis needs. At this stage, we clarify whether the project is focused on whole-genome SV discovery, a candidate region, population comparison, breeding material comparison, strain-level variation, or integration with existing results.
After sample submission, genomic DNA quality is checked before library preparation. For long-read workflows, DNA integrity is especially important because long molecules improve the ability to span repeats, breakpoints, and haplotype blocks. If the sample does not match the planned workflow, we review possible adjustments before moving forward.
Depending on the confirmed strategy, samples move into short-read, long-read, or hybrid sequencing. For long-read projects, the goal is to generate reads that can support SV detection, breakpoint resolution, and phasing where the data allows. Reads are then aligned to the reference genome or used in an assembly-aware workflow when appropriate.
Structural variants are called, filtered, classified, and annotated. Haplotype phasing is performed when the data and study design support it. Results can then be connected to genes, regulatory regions, candidate intervals, population groups, or trait-associated regions. You receive output files and a project report that summarize the analysis logic, key result types, file structure, and visualization-ready outputs.
Sample Requirements and Project Intake Information
Sample quality directly affects long-read SV and haplotype analysis. High-molecular-weight DNA is especially important when the project depends on long-read evidence across repeats, SV breakpoints, or phased regions.
Final sample requirements depend on species, genome size, sample type, platform, and project goal. Before project confirmation, our team reviews the information below and recommends the most suitable workflow.
Sample or input type
What we review
Quality focus
Typical QC checkpoints
Notes
High-molecular-weight genomic DNA for long-read analysis
DNA integrity, concentration, purity, extraction method, sample history
Long DNA fragments, low degradation, low contamination
Qubit, NanoDrop, gel, PFGE or fragment-size review where applicable
Best for projects that rely on long-read evidence across repeats, SV breakpoints, or phased regions
Standard genomic DNA for short-read WGS support
DNA amount, purity, degradation, sample consistency
Stable input quality for library construction
Qubit, NanoDrop, gel check, library QC
Useful when short-read data support cohort-level or hybrid analysis
Existing FASTQ, BAM, CRAM, or VCF files
File format, platform source, sample metadata, reference genome version
File integrity, metadata completeness, compatibility with planned analysis
File integrity check, format check, metadata review
Can support reanalysis, hybrid integration, or downstream interpretation
Tissue, cell, plant, microbial, or environmental material
Sample source, preservation condition, expected DNA quality, extraction feasibility
Suitability for DNA extraction and downstream sequencing
Sample inspection, extraction feasibility review, input QC after extraction
Extraction support may be considered when direct DNA submission is not available
Existing GWAS, QTL, BSA, pan-genome, or population datasets
Study design, group labels, candidate regions, reference version, result format
Compatibility with SV and haplotype interpretation
Metadata review, coordinate system review, result-file review
Helps connect SV and haplotype results with downstream biological questions
Demo Results
Demo results help your team understand what the final analysis may look like before starting the project. These examples show result types, not fixed biological conclusions.
SV landscape summary
This output summarizes deletions, insertions, inversions, duplications, translocations, and CNVs across samples or regions.
Haplotype block and phased variant view
This output shows phased variants across a region, helping you see which variants occur together on the same haplotype.
Integrated candidate-region interpretation
This output combines SV calls, phased variants, gene annotation, and group comparison signals in one region.Demo Results
Demo results help your team understand what the final analysis may look like before starting the project. These examples show result types, not fixed biological conclusions.
SV landscape summary
This output summarizes deletions, insertions, inversions, duplications, translocations, and CNVs across samples or regions.
Haplotype block and phased variant view
This output shows phased variants across a region, helping you see which variants occur together on the same haplotype.
Integrated candidate-region interpretation
This output combines SV calls, phased variants, gene annotation, and group comparison signals in one region.
FAQ
1. What is structural variant and haplotype analysis?
Structural variant and haplotype analysis identifies large genome changes and organizes variants by allele or linked genomic background. It may include SV calling, CNV analysis, breakpoint review, phasing, annotation, visualization, and downstream interpretation.
2. When is SNP-only variant analysis not enough?
SNP-only analysis may be insufficient when the research signal involves large insertions, deletions, inversions, duplications, CNVs, translocations, repeats, or linked allele-specific patterns. If a candidate region looks important but SNPs do not explain the pattern, SV and haplotype analysis may be useful.
3. Why are long reads useful for structural variant detection?
Long reads can span larger genomic regions, repetitive sequences, and variant breakpoints. This makes them useful for detecting and resolving SVs that may be difficult to characterize with short reads alone.
4. How do PacBio and Nanopore differ for SV and haplotype projects?
PacBio-style workflows are often valued for accurate long reads, while Nanopore-style workflows can provide very long reads and strong spanning power. The better choice depends on sample quality, genome complexity, target variant types, read-length needs, and downstream analysis goals.
5. Can this solution work for non-model organisms?
Yes, many non-model organism projects are suitable, but workflow design matters. We review reference genome quality, genome size, repeat content, heterozygosity, ploidy, and sample quality before recommending a strategy.
6. What sample information is needed before recommending a workflow?
We usually need species, sample type, number of samples, available DNA amount, DNA quality, reference genome status, existing sequencing data, target variant types, and the main research question.
7. What deliverables can I expect?
Deliverables may include QC summaries, alignment files, SV callsets, phased variant outputs, annotation tables, visualization-ready files, genome browser tracks, and a project report. Optional outputs can include cohort comparison or candidate-region interpretation.
8. Can SV and haplotype results be integrated with GWAS, QTL-seq, BSA, or pan-genome analysis?
Yes. SV and haplotype results can be linked to mapped intervals, candidate regions, population groups, pan-genome presence/absence patterns, or trait-associated signals when the study design supports it.
9. Do you provide visualization-ready outputs?
Yes. We can prepare summary figures, genome browser tracks, region-level plots, SV class summaries, haplotype block views, and candidate-region panels when these outputs are included in the analysis plan.
10. How should I decide between sequencing-only and a full analysis solution?
Sequencing-only may be enough if your team already has a validated pipeline and clear interpretation plan. A full analysis solution is more useful when you need help with platform selection, SV calling, phasing, annotation, visualization, and downstream biological interpretation.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India