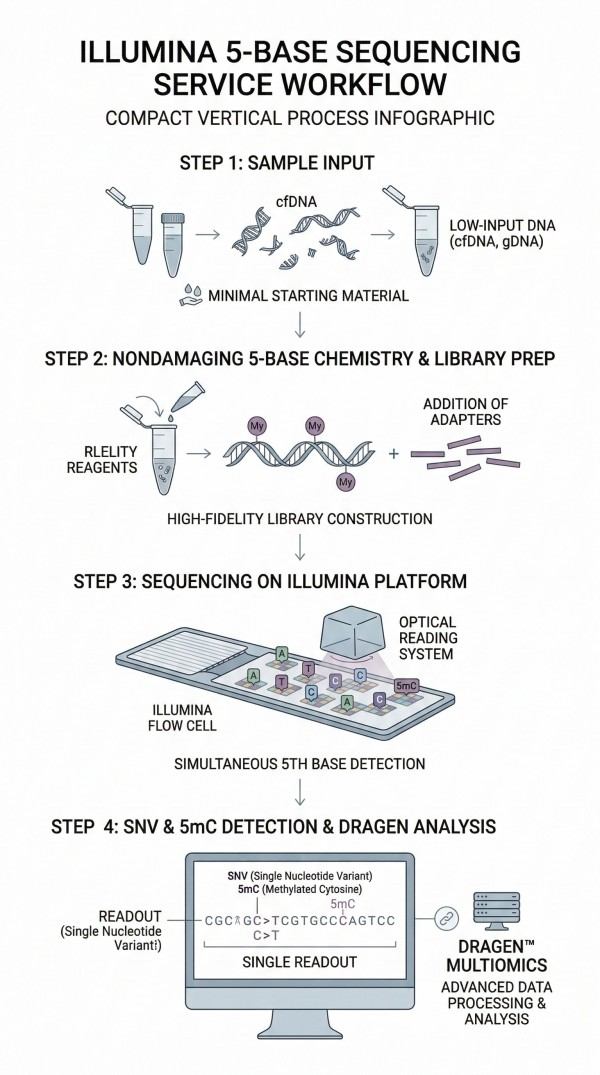
Illumina 5-base Sequencing Service
Home
> Illumina 5-base Sequencing Service
Illumina 5-base Sequencing Service: Unified Insights into the Genome and Methylome
In the rapidly evolving landscape of epigenomics, understanding the intricate relationship between genetic variation and chemical modifications is paramount. DNA is inherently multiomic, carrying not only the genetic code but also modified bases that hold critical epigenetic information. The Illumina 5-base Sequencing Service provided by N2 Jenomics Lab Pvt. Ltd. represents a fundamentally different approach to DNA methylation analysis. By enabling the simultaneous detection of five distinct bases—Adenine (A), Thymine (T), Guanine (G), Cytosine (C), and 5-methylcytosine (5mC)—in a single readout, this service eliminates the need to choose between genomic and epigenomic investigations.
At N2 Jenomics Lab Pvt. Ltd. , we leverage this novel chemistry and optimized bioinformatics algorithms to provide a streamlined Epigenomics workflow that maximizes sequencing output and mapping efficiency. By capturing combined methylome and genome insights with exceptional coverage uniformity, we empower global researchers to make every read count in their quest to decipher complex gene regulation networks and discover next-generation biomarkers.
Simultaneous 5mC and SNV detection in a single readout
Nondamaging chemistry preserving high nucleotide diversity
A Revolution in DNA Methylation Analysis: The 5-Base Chemistry
For decades, the gold standard for detecting DNA methylation has relied on harsh chemical treatments or multi-step enzymatic reactions. Traditional methods, such as Whole Genome Bisulfite Sequencing (WGBS), utilize sodium bisulfite to deaminate unmethylated cytosine into uracil (which is subsequently read as thymine during PCR amplification). While effective for basic methylation calling, this chemical conversion comes with severe analytical penalties. Bisulfite treatment is highly destructive, causing widespread depurination and DNA strand breaks. This results in the loss of up to 90% of the input DNA, severely limiting its application for precious or low-input samples.
Beyond Traditional Conversion: Nondamaging and Direct
The Illumina 5-base solution utilizes a groundbreaking novel chemistry that bypasses the destructive deamination process entirely. Instead, it allows for the direct conversion of 5mC to T in a simple, single-step reaction that is entirely non-damaging to the DNA template.
Because the native DNA backbone remains intact, the original library complexity is preserved. This non-destructive methodology ensures high-quality data generation even from the most challenging biological inputs, circumventing the data gaps and genomic dropouts classically associated with bisulfite sequencing.
Comparison of DNA integrity between 5-base direct conversion and traditional methods.
Maximum Output with High Mapping Efficiency
One of the primary analytical hurdles of traditional bisulfite sequencing is the drastic reduction in nucleotide diversity. When all unmethylated cytosines are converted to thymines, the genome effectively becomes a three-base sequence (A, T, G). This severely impairs the ability of alignment software to accurately map reads back to the reference genome, leading to massive data waste.
The 5-base approach solves this by retaining high nucleotide diversity. Because only the methylated cytosines (5mC) are converted, the resulting sequencing libraries more closely resemble the natural, four-base genomic sequence. This leads to several distinct computational advantages:
Greater Mapping Efficiency: Algorithms can align reads faster and with much higher confidence, drastically reducing the percentage of unmapped or ambiguously mapped reads.
High Coverage Uniformity: The preservation of sequence complexity reduces GC-bias and ensures that traditionally difficult-to-sequence regions of the genome are represented accurately.
Improved Variant Calling: The retained sequence context enables the high-accuracy detection of germline single nucleotide variants (SNVs) directly alongside methylation data, a feat nearly impossible with standard WGBS.
Our end-to-end service is strategically designed to simplify data interpretation and accelerate multiomic discovery. We provide a robust path from sample preparation to actionable, publication-ready insights.
Integrated Library Prep to Analysis
The workflow begins with rigorous sample preparation and library construction, engineered to maintain the highest levels of molecular fidelity.
Broad Sample Compatibility: Our optimized protocols are highly compatible with multiple sample types, including high molecular weight genomic DNA and highly fragmented cell-free DNA (cfDNA).
Workflow Efficiency: The streamlined library preparation bypasses lengthy multi-day enzymatic conversions and can be completed in less than a day, reducing overall turnaround times.
Strategic Flexibility: We support both whole-genome sequencing (WGS) and targeted enrichment strategies, allowing researchers to scale their investigations according to specific project needs and budgetary constraints.
Sequence and Analyze with Confidence
N2 Jenomics Lab Pvt. Ltd. utilizes the latest high-throughput sequencing technology to ensure the highest data quality and statistical power.
Platform Excellence: Sequencing is executed on state-of-the-art systems, including the NovaSeq™ X Series or NextSeq™ 2000 System, guaranteeing massive throughput and exceptional base-calling accuracy.
Advanced DRAGEN™ Analysis: The computational heavy lifting is powered by DRAGEN secondary analysis, enabling high-accuracy dual genomic and epigenomic annotations at unprecedented speeds.
Clear Visualizations: Complex multiomic analysis results are delivered with easy-to-use, clear visualizations through Illumina Connected Multiomics platforms.
High-Accuracy Multiomic Deliverables
The ultimate objective of the Illumina 5-base Sequencing Service is to provide dual, high-resolution insights within the exact same sequencing reads. This localized, richer context is vital for revealing cis-regulatory mechanisms of gene expression and identifying novel biomarkers for complex disease research.
Simultaneous SNV and 5mC Detection
Our service delivers uncompromising accuracy for both genetic and epigenetic features simultaneously.
Methylation Detection Precision: The unique chemistry ensures high-precision (often >99%) and high-sensitivity 5mC calling across the entire genome, accurately capturing the methylation landscape.
Genetic Variant Accuracy: Unlike legacy methods where C-to-T conversions obscure true C>T single nucleotide polymorphisms, the 5-base platform's accuracy for germline SNV detection remains exceptionally high, performing on par with standard, non-converted WGS.
Bioinformatics Analysis & Customization
The bioinformatics pipeline provided by N2 Jenomics Lab Pvt. Ltd. ensures that you receive a comprehensive, structured package of analytical deliverables:
Primary Data & QC: Delivery of raw FASTQ files accompanied by rigorous quality control reports detailing conversion rates, library diversity, and insert size distributions.
Standard Deliverables: Provision of DRAGEN-processed alignment files (BAM/CRAM), comprehensive VCF files for SNVs and Indels, and highly accurate 5mC methylation calls (provided in VCF or BigWig formats for genome browser integration).
Advanced Multiomic Insights: For researchers requiring deeper investigation, our team provides specialized 5mC/5hmC Sequencing Services to differentiate between distinct cytosine modifications, as well as complex Differential Methylation Region (DMR) profiling and pathway enrichment analysis.
Technical Comparison & Sample Requirements
Understanding how 5-base sequencing compares to traditional methods is essential for choosing the right technology for your samples.
Method Comparison Table
Metric
Bisulfite Sequencing
On-market Enzymatic
Illumina 5-base Solution
DNA Damage
High
Medium
Low
Nucleotide Diversity
Low
Low
High
Workflow Complexity
High
High
Low
Methylation Accuracy
High
High
High
Variant Accuracy
Low
Low
High
Sample Requirements Table
To ensure optimal library construction and data generation for your 5-base sequencing project, please adhere to the following stringent sample submission guidelines.
Fragmentation profile (Bioanalyzer or TapeStation)
Reference
The 5-base genome: a simultaneous view of genomics and epigenomics. https://aacrjournals.org/cancerres/article/85/8_Supplement_1/3159/757116/Abstract-3159-The-5-base-genome-a-simultaneous
The 5-base solution: A new approach to DNA methylation. https://www.illumina.com/science/genomics-research/articles/5-base-solution.html
Illumina 5-Base DNA Prep Product Page. https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/illumina-5-base-dna-prep.html
Illumina Technical Flyer: 5-base solution for methylation and variant detection. https://www.illumina.com/content/dam/illumina/gcs/assembled-assets/marketing-literature/illumina-5-base-methylation-flyer-m-gl-03401/illumina-5-base-methylation-flyer-m-gl-03401.pdf
Compliance & Disclaimer: For Research Use Only. Not for use in diagnostic procedures.
Demo Results
Visualization of Multiomic Deliverables. The retained sequence context enables the high-accuracy detection of germline single nucleotide variants (SNVs) directly alongside methylation data.
5-base Sequencing Service FAQs
1. How does the 5-base solution fundamentally differ from traditional Whole Genome Bisulfite Sequencing (WGBS)?
WGBS employs harsh sodium bisulfite chemicals to deaminate unmethylated cytosines into uracil. This process significantly damages the DNA backbone (causing fragmentation) and drastically reduces sequence diversity, leading to poor mapping efficiency. The 5-base solution, conversely, uses a proprietary chemistry to directly convert only 5mC to T. This preserves DNA integrity, maintains a high-diversity library, and allows for simultaneous, high-accuracy variant calling and methylation detection.
2. Is this sequencing service suitable for highly fragmented cell-free DNA (cfDNA) samples?
Yes. The Illumina 5-base solution is exceptionally compatible with highly fragmented and limited sample types, including cfDNA isolated from liquid biopsies. Because the chemistry is non-destructive and does not induce further strand breaks, it is particularly well-suited for low-input oncology or non-invasive prenatal testing (NIPT) research models where DNA preservation is absolutely critical.
3. What specific bioinformatics outputs and file formats are provided upon project completion?
Clients receive a comprehensive data package. This includes raw sequencing data (FASTQ files) for archival purposes, along with processed files generated by the DRAGEN secondary analysis pipeline. The processed deliverables feature dual annotations, providing BAM/CRAM files for alignments, standard VCF files for SNVs and Indels, and specialized VCF or BigWig files detailing the 5mC methylation status—all derived from the same foundational sequencing reads.
Case Study: Validating Dual-Omics at the RAMP1 Locus
Validating Simultaneous Genomic and Epigenomic Annotations
Background
Investigating Allele-Specific Methylation (ASM) and cis-regulatory elements requires exact knowledge of both the genetic variant and the methylation status on the same DNA molecule. Historically, researchers had to perform WGS for variant calling and WGBS for methylation, then attempt to correlate the two statistically—a method fraught with noise and alignment errors. To demonstrate the transformative power of simultaneous genome and methylome sequencing, this validation study focuses on the RAMP1 (Receptor Activity-Modifying Protein 1) locus on Chromosome 2.
Materials & Methods
Sequencing Protocol
Utilizing the Illumina 5-base chemistry for single-step 5mC conversion.
Sequencing on high-throughput NovaSeq platforms.
Bioinformatics Analysis
DRAGEN algorithms mapping reads covering genomic coordinates 237,855,000 to 237,915,000 on Chromosome 2.
Results and Multiomic View
Figure: Dual detection of SNV and methylation at the RAMP1 locus on Chromosome 2.
Variant Detection: The sequencing output successfully identified a heterozygous A>C variant within the RAMP1 gene architecture.
Methylation Profiling: Because the method preserves nucleotide diversity and does not damage the DNA, the DRAGEN algorithms mapped reads with extremely high confidence. Within the exact same read tracks containing the SNVs, the data clearly distinguished between methylated and unmethylated alleles at adjacent CpG sites.
Conclusion
This validation proves that the 5-base solution can directly observe the interplay between a specific genetic mutation and its localized epigenetic state. By capturing both data points simultaneously, researchers can definitively link genetic variants to epigenetic regulatory changes, accelerating discoveries in oncology, rare genetic diseases, and developmental biology.
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India