Long Amplicon Analysis (LAA) FAQs
1. What are the main application areas of Long Amplicon Analysis?
Long Amplicon Analysis finds extensive application in an array of research areas, which include:
- Studies on Genomic Structural Variations: It is employed for analyzing intricate variations encompassing significant insertions, deletions, inversions, and repeated sequences.
- Research on Genetic Diseases: This tool aids in the identification of complex mutations that involve elongated genomic regions associated with clinical diagnosis.
- Studies on Evolution and Phylogeny: Long Amplicon Analysis allows exploration of genetic variations between species, thereby shedding light on evolutionary relationships.
- Research on Microbial Diversity: It is used to investigate the structure of the whole genome of microbial communities in various environmental samples.
2. What is the basic workflow of Long Amplicon Analysis?
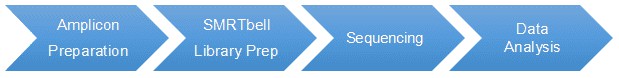
The core processes that underpin Long Amplicon Analysis commence with DNA extraction and cleansing. The operation entails isolating optimum quality DNA from the given samples. This is followed by a PCR amplification process where specific primers are used in conjunction with high-fidelity polymerases to intensify target segments.
Subsequently, the amplicon purification process takes place, in which residual unamplified DNA and primer dimers are eliminated. This leads to the step of library preparation and a quality control check. Here, libraries compatible with sequencing platforms are prepared and a rigorous quality check is conducted to ensure their suitability for subsequent procedures.
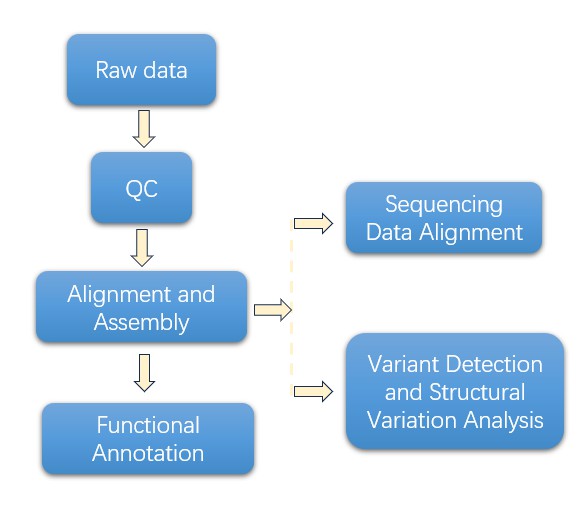
High-throughput sequencing is the next key stage which makes use of platforms like PacBio and Oxford Nanopore adept for long-read sequencing. Following the sequencing step, a comprehensive data analysis is performed. This analysis encompasses quality control, alignment, variant detection, and functional annotation.
Before the data is stored and made available for distribution, there is an important stage of result validation and reporting. Key findings are experimentally verified, and comprehensive analysis reports are generated. As a concluding step, the data is securely stored in databases, following which it is made accessible for sharing in line with the research requirements.
3. What are the commonly used tools in Long Amplicon Analysis data analysis?
Tools used in data analysis include:
Quality control: FastQC, Trimmomatic, Cutadapt.
Alignment and assembly: BWA, Bowtie2, Minimap2, Canu, Flye.
Variant detection: GATK, FreeBayes, Manta, LUMPY.
Functional annotation: ANNOVAR, SnpEff.
4. How to ensure the accuracy and reliability of Long Amplicon Analysis results?
Several methodological protocols can be put in place to bolster accuracy and reliability, such as:
- DNA Extraction and Purification: The implementation of sophisticated kits coupled with meticulous procedure execution can facilitate high-quality DNA extraction and purification.
- PCR Conditions Optimization: Fine-tuning specific conditions within the polymerase chain reaction (PCR) process, such as annealing temperature and extension time, can help secure efficient segment amplification.
- High-Fidelity Polymerase Utilization: The application of high-fidelity polymerases plays an important part in minimizing chances of amplification discrepancies.
- Attentive Quality Control: Stringent quality control measures should be observed at critical junctions during the sequencing and data analysis phases.
- Experimental Validation: Pivotal results should be validated experimentally. This could involve methods such as Sanger sequencing or quantitative PCR (qPCR).