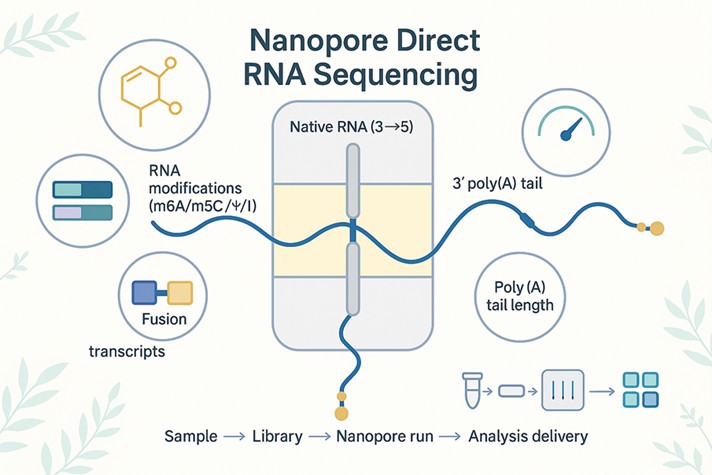
Nanopore Direct RNA Sequencing is the only approach that reads native RNA molecules directly—without reverse transcription or PCR—so your data preserve real-world base modifications and per-molecule features. Sequencing proceeds 3′→5′ from a poly(T) adapter, producing single-molecule long reads that span complete 5′-UTR–CDS–3′-UTR structures.
How it works
Enrich & adapt: Poly(A)+ RNA (or a custom capture) is prepared and ligated to a poly(T) adapter with a motor protein.
Translocate through the pore: Each RNA strand is ratcheted through a nanopore where ionic current changes encode nucleotide identities.
Decode & analyze: Basecalling reconstructs sequence while signal-aware algorithms output RNA modification signatures (m6A/m5C/Ψ/I) and poly(A) tail length per read.
Orientation and chemistry are documented in our validated Direct RNA sequencing Nanopore protocol as part of the Nanopore direct RNA sequencing workflow.
Why it matters
Full-length isoforms, no assembly: Long reads directly resolve alternative splicing and complex transcript structures, reducing inference errors common to short-read RNA-seq.
Native epitranscriptome: Because there is no RT/PCR, true chemical RNA modifications are retained and detectable at near base-level resolution.
Poly(A) biology: Per-read poly(A) tail length enables studies of stability, translation, and isoform-specific regulation.
Lower amplification/GC bias: PCR-free libraries improve quantification of GC-extreme and structured RNAs.
Nanopore Direct RNA Sequencing Workflow
1. Sample receipt & QC
Accepted input: total RNA (recommended ≥50 µg at ≥180 ng/µL; workable 20–80 µg, study-dependent).
Integrity check (Bioanalyzer/TapeStation), contaminants screen, rRNA content; RNase-free handling.
Optional controls: ERCC spike-ins; methylation standards for caller calibration.
Recommendation: ≥2 biological replicates per condition (≥3 improves power).
2. Poly(A)+ selection / custom capture
Standard: oligo-dT enrichment for mRNA.
Options: rRNA depletion or targeted capture (e.g., lncRNA/circRNA panels or organism-specific probes).
3. Library preparation — Direct RNA 1D (PCR-free)
Ligation of poly(T) adapter and motor protein; no RT / no PCR.
Orientation is 3′→5′ by design; chemistry and barcoding are documented in our validated Direct RNA sequencing Nanopore protocol.
4. Nanopore run & real-time monitoring
Flow cell QC, pore occupancy, live yield tracking.
Run configuration matched to depth targets (organism size, complexity, comparisons).
5. Basecalling & signal processing
Raw FAST5 → FASTQ with quality metrics.
Signal-aware extraction of per-read poly(A) tail length and modification features (m6A/m5C/Ψ/I candidates).
6. Alignment & isoform handling
Reference-guided spliced alignment.
Isoform clustering & de-redundancy to mitigate 5′ truncation and molecule multiplicity; poly(A) site usage captured.
7. Primary analytics hand-off
Structured outputs (BAM/CRAM, GTF/GFF, per-read poly(A) tables, modification feature tables) passed into the Nanopore direct RNA sequencing analysis modules.
Notes & best practices
PCR-free libraries reduce GC/amp bias and preserve native RNA modifications.
Some 5′ truncation is expected; our clustering pipeline ensures isoform-level accuracy.
Optional nascent RNA module: 5-EU labeling enables identification of nascent transcripts, half-life estimation, and stability analyses.
Integrated Analysis Plan
Nanopore Direct RNA Sequencing Bioinformatics Analysis
I. Isoform analysis
Alternative splicing analysis
Fusion transcript identification
Poly(A) tail analysis
II. Expression quantification
Transcript quantification
Differential transcript analysis
Functional enrichment (GO/KEGG/GSEA)
Protein–protein interaction (PPI) network
III. RNA methylation / modification analysis
m6A site annotation
Modification enrichment analysis
Differential m6A analysis
IV. Isoform joint quantitative expression analysis
AltTP analysis
Functional Diversity Analysis (FDA)
Differential enrichment analysis
V. Nascent mRNA analysis (optional)
Nascent mRNA identification
Half-life estimation
mRNA stability correlation analysis
Differential nascent expression
What You'll Get from Our Nanopore Direct RNA Sequencing Service
Comprehensive Direct RNA Outputs
Fully processed datasets including FAST5/FASTQ, spliced BAM/CRAM, GTF/GFF (novel/known isoforms, TSS/TTS, poly(A) sites), poly(A) tail tables, RNA modification site lists (m6A/m5C/Ψ/I), fusion calls, and transcript-level counts/TPM—ready for immediate Nanopore direct RNA sequencing analysis and downstream use.
Differential & Functional Insights
Condition-specific results spanning differential transcripts, AltTP/DIU, differential m6A, and poly(A) comparisons, plus GO/KEGG enrichment and optional PPI network overlays to link isoform/modification changes with pathways.
Publication-Ready Visualizations
High-quality figures: isoform heatmaps, volcano plots, sashimi/AltTP/DIU visuals, fusion breakpoint panels, modification metagene/motif views, and poly(A) histograms—optimized for interpretation and journal submission.
Transparent Analysis Records
A concise Methods & QC report (Direct RNA 3′→5′ protocol summary, software/versions/parameters, yield/read-Q/mapping/junction metrics) with pipeline manifest, environment file, and checksums for full reproducibility.
Optional Add-Ons
Nascent RNA module (5-EU identification, half-life, stability correlations), integrative multi-omics (e.g., Illumina depth + Direct RNA features), non-poly(A) capture strategies, interactive HTML dashboards, and custom exports (e.g., Cytoscape-ready).
Sample Requirements & Shipping
Category
Requirements & Notes
Standard Input
Total RNA: recommended ≥50 µg at ≥180 ng/µL; workable 20–80 µg (aim-dependent). Cell guide: ~1×10^7 cells typically yield sufficient RNA. RNA type: Poly(A)+ mRNA by default; non-poly(A) options available (custom capture/NERD-like).
Quality Criteria
Integrity: RIN ≥7 or high DV200. Cleanliness: avoid phenol/EDTA carryover and DNase inhibitors.
Replicates & Controls
Replicates: ≥2 biological per condition (≥3 preferred for differential/AltTP). Controls (optional): ERCC spike-ins, methylation standards (for modification caller calibration).
Submission & Shipping
Packaging: RNase-free tubes, clearly labeled (project ID, sample ID, concentration, volume). Temperature: ship on dry ice. Manifest: include sample metadata (organism, treatment, intended comparisons). Compliance: follow international biospecimen regulations; include SDS if required.
Low-Input / Special Cases
Consult us for low-input RNA strategies, targeted capture panels (lncRNA/circRNA), or degraded/archival tissues (case-by-case feasibility).
Applications & Case Snapshots
Core applications
Isoform-resolved transcriptomics: full-length isoforms, alternative splicing (AltTP/DIU), TSS/TTS & poly(A) site usage.
Fusion transcript detection: single-molecule evidence across breakpoints for high-confidence calls.
Epitranscriptomics: transcriptome-wide RNA modification mapping (m6A/m5C/Ψ/I) with differential analysis.
Poly(A) tail biology: per-read poly(A) tail length to study stability and translation; isoform-specific differences.
1. Native-RNA expertise Direct RNA 1D, PCR-free, 3′→5′ orientation; validated Direct RNA sequencing Nanopore protocol with rigorous sample/run QC.
2. End-to-end workflow & analysis From sample QC and library prep to sequencing, Nanopore direct RNA sequencing analysis, and integrated reporting—isoforms, m6A/m5C/Ψ/I, poly(A) tails, and optional nascent RNA.
3. Proven in peer-reviewed research Nanopore direct RNA libraries prepared by N2 Jenomics Lab Pvt. Ltd. were used in a PLOS Genetics study on Arabidopsis epitranscriptomics, demonstrating single-molecule m6A mapping with DRS (Sun et al., 2022).
4. Quality & reproducibility Transparent Methods & QC report, version-locked pipelines, manifests and checksums; publication-ready figures and reusable data packages.
5. Designed for your question Study design for isoforms/splicing, epitranscriptome, poly(A) biology, nascent RNA; options for non-poly(A) capture and multi-platform integration.
Demo Results
Partial results are shown below:
Differential Isoform Expression Heatmap
Distribution of m6A Sites across Transcript Region
Differential Isoform Volcano Plot
m6A Sequence Motif Logo
Sequence Coverage at a Gene Locus
Nanopore Direct RNA Seq FAQs
Q1. When is Direct RNA not the first choice for analysis?
For large, gene-level differential expression screens under tight budgets, cDNA/Illumina usually offers more depth per dollar. Direct RNA's throughput is lower and cost per usable read is higher, so it's best reserved for questions where native-RNA features matter—full-length isoforms, alternative splicing, fusion transcripts, RNA modifications, and poly(A) tail biology.
Q2. How does Nanopore Direct RNA Sequencing differ from traditional RNA-seq?
Traditional RNA-seq converts RNA to cDNA and amplifies it by PCR, which can introduce bias and remove native chemical marks. Nanopore Direct RNA reads native RNA molecules directly in a 3′→5′ orientation without RT/PCR, preserving RNA modifications and enabling per-read poly(A) tail measurements alongside full-length isoforms.
Q3. What are the main challenges of Direct RNA?
Direct RNA currently has lower throughput than cDNA and higher single-read error than Illumina, and it requires specialized, signal-aware analysis and adequate compute. In practice, we mitigate these limitations with appropriate experimental design, consensus across reads, robust alignment, and careful QC.
Q4. Can Direct RNA do gene expression, splicing isoform, and fusion transcript analysis?
Yes, provided the study is designed for these endpoints. We routinely quantify transcripts, analyze isoform usage (AltTP/DIU), and detect fusion transcripts with single-molecule evidence; however, for very large cohorts focused solely on gene-level DE, cDNA/Illumina can be more economical, while Direct RNA is favored for mechanism-driven studies where native-RNA features are critical.
Q5. Can poly(A) tails and RNA modifications be profiled in the same run?
Yes. Both poly(A) tail length and modification signatures such as m6A, m5C, and Ψ/inosine are derived from the same nanopore signal on native RNA, enabling joint interpretation on the very molecules that carry the features.
Q6. Is Direct RNA strand-specific, and what about 5′ truncation?
Reads are inherently strand-specific and proceed 3′→5′ from a poly(T) adapter. Some reads are 5′-truncated by chemistry and pore dynamics; our isoform clustering and de-redundancy steps account for this so that isoform-level calls remain reliable.
Q7. What input do I need?
We recommend ≥50 µg total RNA at ≥180 ng/µL, with a workable range of 20–80 µg depending on goals and quality; intact RNA (e.g., RIN ≥ 7 or high DV200) markedly improves outcomes. See Sample Requirements for shipping and QC details.
Q8. Do Nanopore Direct RNA reads detect m6A directly?
Yes. Because native RNA passes the pore without RT/PCR, m6A produces characteristic current signatures that can be called at near base-level resolution and linked to specific transcripts and isoforms.
Q9. How much RNA is needed for ONT Direct RNA sequencing?
Most projects target ≥50 µg total RNA at ≥180 ng/µL; studies with 20–80 µg can be feasible depending on aims and RNA integrity, which strongly influences yield and analysis depth.
Q10. How does Direct RNA differ from cDNA long-read or Illumina short-read RNA-seq?
Direct RNA reads native RNA (3′→5′) with no RT/PCR, preserving RNA modifications and enabling per-read poly(A). cDNA/short-read lose native modifications and infer isoforms from fragments.
Nanopore Direct RNA Seq Case Studies
Customer Publication Highlight
FIONA1-mediated methylation of the 3'UTR of FLC affects FLC transcript levels and flowering in Arabidopsis
Journal: PLoS Genetics Author: Sun, B., Bhati, K. K., Song, P., et al. Published: September 27, 2022
1) Background
Flowering time in Arabidopsis thaliana is tightly controlled by RNA regulatory layers, including N6-methyladenosine (m6A). Sun et al. investigated whether the m6A writer FIONA1 (FIO1; METTL16-like) adds methyl marks to the 3′UTR of FLC and thereby stabilizes the transcript. To obtain an unbiased, single-molecule view of RNA methylation and isoform coverage, the team integrated Illumina RNA-seq, MeRIP-seq, and Nanopore Direct RNA Sequencing (DRS).
Library prep: ONT SQK-RNA002 Direct RNA protocol; libraries prepared by N2 Jenomics Lab Pvt. Ltd. .
Sequencing: PromethION (R9.4), ~48–72 h runs.
Basecalling/QC: Guppy v3.2.6; filtering with NanoFilt.
Alignment & correction: minimap2 mapping; Fclmr2 correction; metrics via samtools.
m6A calling & stats: Tombo (de novo) + MINES; differential methylation with methylKit.
3) Results
Nanopore direct RNA sequencing analysis revealed genotype-dependent transcriptome changes and an Arabidopsis m6A consensus of RGACH. In wild type, most m6A events occurred at GGACA (34.7%), followed by AGACT (27.2%), GGACT (22.9%), and GGACC (15.2%). Across fio1 mutants, DRS identified 74 hypomethylated genes (fio1-1) and 63 (fio1-5) relative to WT. Critically, DRS coverage confirmed depletion of FLC mRNA in fio1-1 and fio1-5, consistent with loss of 3′UTR methylation.
4) Conclusions
This study demonstrates how Nanopore Direct RNA Sequencing provides single-molecule, full-length evidence linking 3′UTR m6A to mRNA stability. By preserving native RNA, DRS directly couples m6A site detection, motif discovery, and isoform-level coverage—resolving that FIO1 installs m6A at the FLC 3′UTR, and that loss of this mark leads to rapid FLC decay and early flowering.
Direct RNA-sequencing analysis.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India