Nanopore Ultra-Long Sequencing
Home
> Nanopore Ultra-Long Sequencing
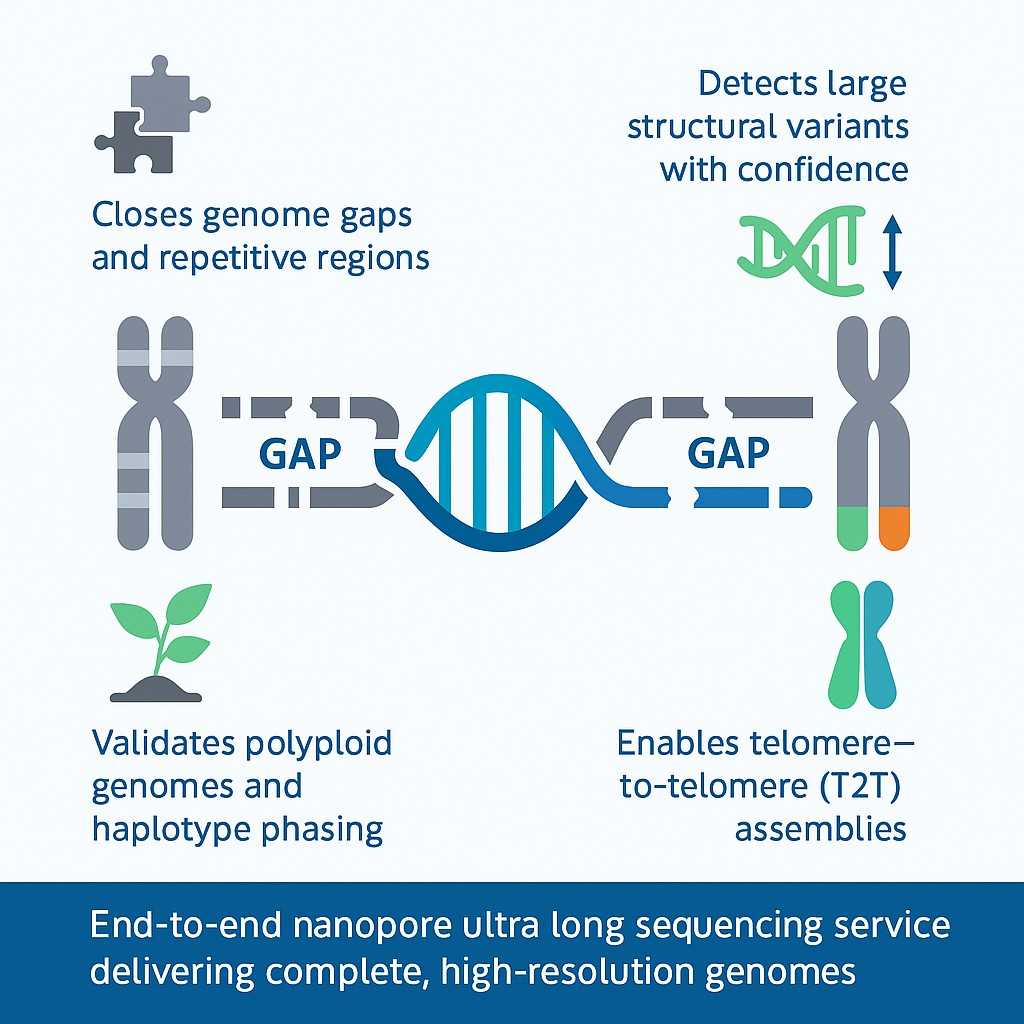
Nanopore Ultra-Long Sequencing Service for Complete Genome Assemblies
N2 Jenomics Lab Pvt. Ltd. provides advanced nanopore ultra long sequencing solutions designed for researchers tackling complex genomes. Using the latest Nanopore Ultra-Long Sequencing Kit, our workflow generates reads exceeding 100 kb (N50), with maximum read lengths surpassing 4 Mb. This approach enables high-contiguity assemblies, resolves repetitive regions, and supports telomere-to-telomere genome research.
Our service is built for research teams in agriculture, biotechnology, and drug development who require gap-free assemblies and structural variant analysis. By combining optimized DNA extraction, proven library preparation, and bioinformatics expertise, N2 Jenomics Lab Pvt. Ltd. delivers reliable data for high-impact projects.
Key Advantages:
Ultra long DNA sequencing nanopore closes gaps in repetitive and polyploid genomes
Nanopore long read sequencing achieves read lengths beyond 100 kb
Complete end-to-end support, from DNA preparation to analysis
Conventional sequencing often leaves unresolved gaps, particularly in genomes with extensive repeats, polyploidy, or high structural complexity. These gaps limit the accuracy of genome assemblies and reduce confidence in downstream analyses.
Nanopore ultra long sequencing addresses these limitations by generating DNA reads far longer than standard approaches. Unlike short- or mid-length sequencing, which struggles with highly repetitive regions, ultra long reads can span centromeres, telomeres, and structural variants in a single stretch.
This capability has transformed genome research. Plant and animal studies now routinely achieve telomere-to-telomere (T2T) assemblies, enabling researchers to explore genetic architecture with unprecedented resolution. For applied research in crop breeding, biomedical discovery, and microbial evolution, the ability to resolve complete genomes provides a direct competitive advantage.
Whole genome sequencing uncovers virulence factors, mobile genetic elements, and potential environmental transmission of bacterial strains in cattle farm settings. (Rivu, Supantha, et al., 2024)
Technical Parameters
Parameter
Specification
Read length
N50 >50–100 kb; maximum reads >4 Mb
Input requirement
≥6 million cells (PBMCs, cultured cells, or frozen tissue)
Qubit, Nanodrop, pulsed-field gel electrophoresis (PFGE)
Storage & logistics
Kits shipped at 2–8 °C; long-term storage at –20 °C
Advantage: N2 Jenomics Lab Pvt. Ltd. Service Highlights
N2 Jenomics Lab Pvt. Ltd. delivers an end-to-end nanopore ultra long sequencing service that combines advanced laboratory protocols with proven sequencing platforms. Our approach is designed to maximise read length, stability, and accuracy, enabling researchers to close gaps and produce highly contiguous assemblies.
Key Service Advantages
Gapless assemblies: Ultra long DNA sequencing nanopore spans repetitive and GC-rich regions, eliminating assembly gaps.
Structural variant discovery: Long reads detect large insertions, deletions, inversions, and repeat expansions with confidence.
ONT ultra long sequencing only (no Illumina/PacBio)
Completed telomere-to-telomere assembly; validated centromeres and telomeres
Key Applications
De novo genome assembly
Ultra long reads span repetitive elements and GC-rich regions, closing gaps that remain unresolved with short-read or standard long-read sequencing.
Structural variation detection
Nanopore long read sequencing identifies large insertions, deletions, inversions, and repeat expansions that influence genome stability and phenotype.
Polyploid genome analysis
Ultra long DNA sequencing nanopore improves haplotype phasing and validates assemblies in polyploid plants and hybrid species.
Telomere-to-telomere (T2T) assembly
Complete chromosome-level assemblies are achieved by spanning telomeres, centromeres, and rDNA regions with single reads.
Transcriptome and RNA sequencing integration
Combined with Nanopore Direct RNA Sequencing and Nanopore Full-Length Transcript Sequencing, researchers can link genome structure with transcriptional activity.
Workflow: End-to-End Service
N2 Jenomics Lab Pvt. Ltd. offers a complete workflow for nanopore ultra long sequencing, from sample preparation to final data delivery. Each stage is optimised to preserve ultra-high molecular weight DNA and maximise read length.
1. Sample preparation
Support for plant, animal, and microbial samples
Specialised protocols to minimise degradation and remove polysaccharides or secondary metabolites
2. DNA extraction and QC
Extraction of ultra-high molecular weight (uHMW) DNA
Quality checks using Qubit, Nanodrop, and pulsed-field gel electrophoresis
3. Library preparation
Nanopore Ultra-Long Sequencing Kit (SQK-ULK114) with Kit 14 chemistry
Transposase-based fragmentation and rapid adapter ligation
Overnight elution to preserve long DNA molecules
4. Sequencing
Performed on PromethION or GridION platforms
Achieve read lengths >100 kb N50, with maximum reads exceeding 4 Mb
5. Bioinformatics analysis
Real-time basecalling and quality filtering
Assembly, polishing, and variant detection
Optional telomere-to-telomere (T2T) assembly support
6. Reporting and delivery
FASTQ and BAM files with QC metrics
Customised analysis reports for genome assembly or structural variant studies
Bioinformatics Analysis
Basic Analysis
Analysis Stage
Description
Basecalling
Converts raw electrical signals into DNA/RNA sequences using models like Dorado for improved accuracy.
Quality Control (QC)
Includes read length distribution, coverage metrics, and quality scores.
De novo Assembly
Builds high-contiguity genomic assemblies using tools optimized for long reads (e.g., Flye, Canu) .
Polishing & Error Correction
Refines assembly error profiles using long-read self-correction or hybrid polishing workflows .
Advanced Analysis
Analysis Stage
Description
Structural Variant Calling
Detects large indels, duplications, inversions using tools such as Sniffles or CuteSV.
Variant Phasing & SV Annotation
Phases variants across long contigs and annotates SVs for biological interpretation.
Telomere-to-Telomere (T2T) Support
Completes chromosome-level assemblies by closing gaps at repeats, centromeres, and telomeres.
Epigenetic & Base Modification Detection
Detects DNA/RNA modifications (e.g., 5mC, m6A) using Remora or Megalodon during basecalling.
Metagenomic / Taxonomic Classification
Classifies reads in mixed samples using workflows like EPI2ME meta pipelines.
Deliverables
Clients will receive:
Raw sequencing data (FASTQ format)
Quality control report (read length distribution, N50, yield, accuracy)
Optional genome assembly and variant analysis results
Project summary report
Sample Requirements for Nanopore Ultra-long Sequencing
To ensure optimal sequencing results, we require the following sample conditions:
Sample Type
Tissue Type
Requirement (per cell)
Remarks
Animal
Mammalian Blood
≥5 mL
Avoid tube breakage: Use plastic anticoagulant tubes
Least effective sample type,Difficult to achieve N50 >100 Kb
Muscle
≥3 g
Plant
Young Tender Leaves
≥3 g
Preferably from newly germinated seeds
Decontamination protocol:
1. Rinse with 75% ethanol 2. Rinse with sterile water 3. Dry and flash-freeze
Our team provides guidelines on sample preparation to help you achieve the best results.
Demo Results
Read length distribution
The accompanying chart (from Oxford Nanopore Technologies) illustrates how ultra-long sequencing dramatically extends read length compared to standard ligation methods—producing a smooth tail reaching several hundred kilobases.
Wheat genome impact
Incorporating ultra-long ONT reads increased the contig N50 from 341 kb to 2.15 Mb, leading to a near-gapless assembly ideal for downstream genomic research and breeding programs.
Plant T2T advancement
In recent plant genome projects, optimized extraction and library protocols delivered ultra-long reads with an N50 up to 440 kb, significantly enhancing automated telomere-to-telomere (T2T) assembly.
Sorghum assembly success
A complete T2T sorghum genome was assembled using only ONT ultra-long sequencing, demonstrating the method's capability to resolve full chromosomes without needing complementary technologies.
Nanopore Ultra-Long Seq FAQs
Q: What read length can I expect from nanopore ultra-long sequencing?
You can expect N50 read lengths typically ranging from 50 to over 100 kb, and in optimal conditions individual reads can exceed 4 Mb—ultra-long reads enable resolution of complex genomic regions like repeats and centromeres.
Q: Why does ultra-long read length matter for my genome assembly?
Ultra-long reads span highly repetitive or structurally complex regions, enabling gapless or near gapless assemblies such as T2T genomes and improving detection of structural variants that shorter reads cannot resolve .
Q: What impact do long reads have on structural variant detection?
Long nanopore reads improve the discovery of large insertions, deletions, inversions, and repeat expansions by spanning them directly, which simplifies variant calling and reduces ambiguity.
Q: Can nanopore sequencing handle both DNA and RNA samples?
Yes, nanopore sequencing supports direct sequencing of both DNA and RNA molecules without the need for amplification or labelling, which allows you to study transcripts and base modifications alongside genome structure.
Q: What factors affect sample quality for ultra-long reads?
DNA purity and fragment length are critical. High-quality extraction with minimal degradation is essential, and multiple extraction attempts may be needed to obtain ultra-high molecular weight DNA suitable for ultra-long read sequencing.
Q: Is nanopore technology suitable for field or portable applications?
Yes, because nanopore sequencers can process native DNA or RNA in real time in scalable formats—from portable MinION devices to high-throughput platforms—this flexibility supports lab, field, and remote applications
Nanopore Ultra-Long Seq Case Studies
1. Background
The human genome is ~3.1 Gb in size and contains extensive repetitive regions, segmental duplications, and heterozygosity, making it challenging to assemble using short-read sequencing. Conventional technologies fail to resolve centromeres, telomeres, and structural variants, leaving persistent gaps in reference genomes. This case study explores how nanopore ultra long sequencing can overcome these barriers.
2. Methods
Researchers sequenced the GM12878 human cell line (Utah/CEPH pedigree) on the Oxford Nanopore MinION platform with R9.4 1D chemistry. DNA preparation protocols were designed to minimise shearing and preserve ultra-high molecular weight fragments.
Data generated: 91.2 Gb of sequence (~30× coverage) from 39 flow cells
Ultra-long reads: N50 >100 kb; maximum read length 882 kb
Assembly tool: Canu assembler with Illumina short-read polishing for accuracy improvement.
3. Results
Nanopore-only assembly NG50 = ~3 Mb
Adding 5× ultra-long coverage doubled NG50 to 6.4 Mb
Major histocompatibility complex (MHC) (4 Mb) resolved in a single contig
12 large gaps (>50 kb) in GRCh38 were closed
Final accuracy after polishing = 99.88%
Ultra-long reads enabled haplotype phasing across the entire MHC locus.
Chromosome plot illustrating how nanopore ultra long sequencing closed 12 gaps in GRCh38, including the 16 Mb MHC locus. Continuous colour blocks represent contiguous assembly, while white gaps indicate unresolved regions.
4. Conclusions
This study demonstrates that nanopore ultra long read sequencing enables highly contiguous human genome assemblies. The technology resolved complex loci, closed reference gaps, and provided haplotype phasing at chromosome scale. These results highlight its potential for producing near-complete telomere-to-telomere (T2T) assemblies and advancing both fundamental genomics and translational applications.
References:
Lu D, Liu C, Ji W, Xia R, Li S, Liu Y, Liu N, Liu Y, Deng XW, Li B. Nanopore ultra-long sequencing and adaptive sampling spur plant complete telomere-to-telomere genome assembly. Mol Plant. 2024 Nov 4;17(11):1773-1786. doi: 10.1016/j.molp.2024.10.008. Epub 2024 Oct 16. PMID: 39420560.
Prall, T.M., Neumann, E.K., Karl, J.A. et al. Consistent ultra-long DNA sequencing with automated slow pipetting. BMC Genomics 22, 182 (2021).
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O'Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018 Apr;36(4):338-345. doi: 10.1038/nbt.4060. Epub 2018 Jan 29. PMID: 29431738; PMCID: PMC5889714.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India