TAIL Iso-seq FAQs
What is TAIL Iso-seq and how does it differ from regular Iso-seq or RNA-seq?
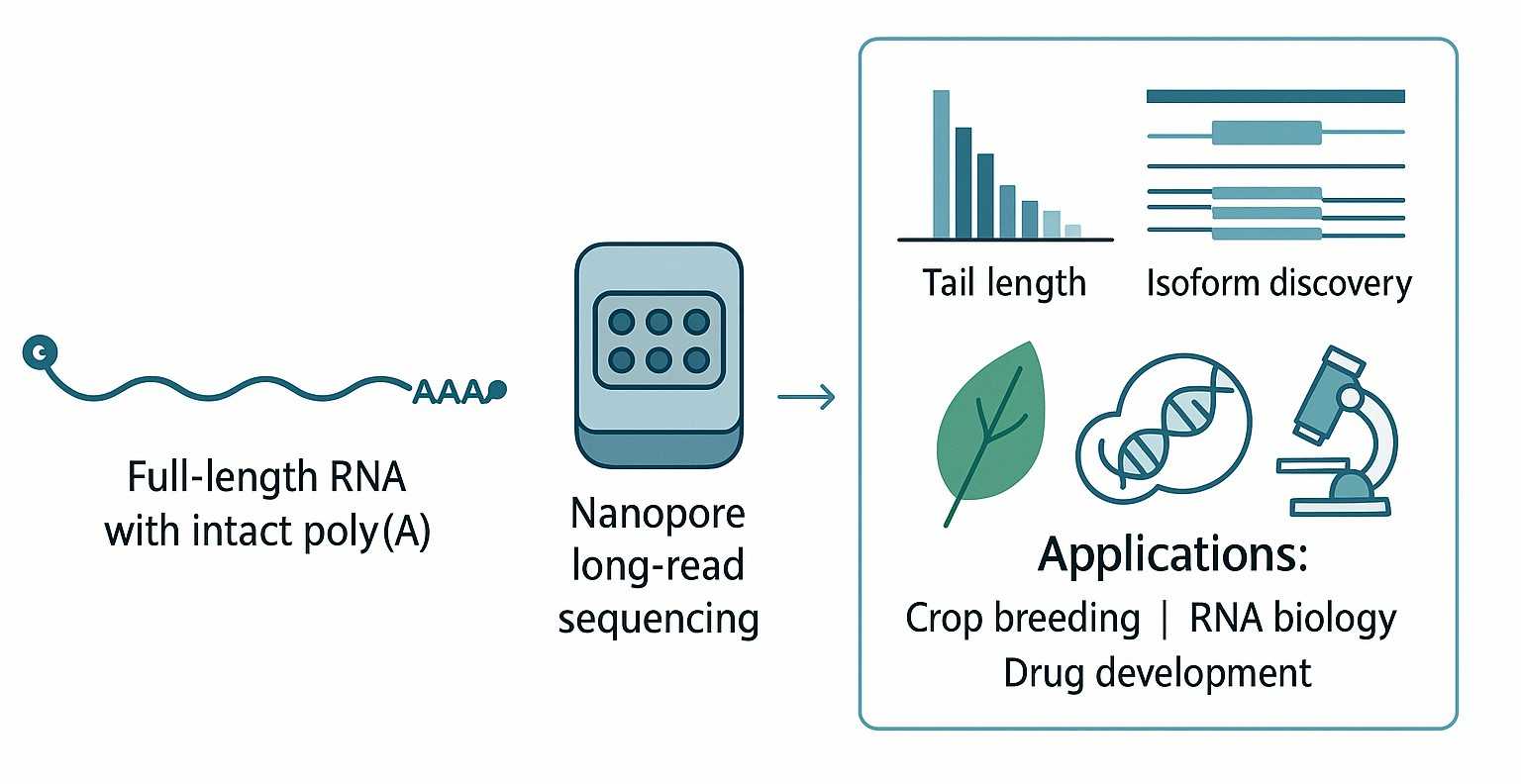
TAIL Iso-seq is a long-read sequencing service (Nanopore-based) that captures full-length transcripts including the native poly(A) tail, enabling direct measurement of tail length, APA (alternative polyadenylation) sites, and isoform diversity. Regular RNA-seq often loses tail information and requires read assembly, while standard Iso-seq may capture transcript structure but not always the full poly(A) composition and non-A residues.
Can TAIL Iso-seq detect non-A nucleotides inside or at the end of poly(A) tails?
Yes. The service includes detection of non-adenosine residues (such as U, G, C) both at the terminal position and internally in the poly(A) tail. These non-A residues provide insight into transcript stability, degradation, or deadenylation regulation. Tools like tailfindr, nanopolish, and others support this analysis.
How accurate is poly(A) tail length measurement with Nanopore long reads?
TAIL Iso-seq uses state-of-the-art basecalling and signal processing pipelines to estimate tail length per molecule with high resolution, often at single nucleotide precision for shorter tails and good relative accuracy for longer tails. Accuracy also depends on sample quality, sequencing output (.fast5 retention), and tool choice (tailfindr, nanopolish, etc.).
Do I need a reference genome to use TAIL Iso-seq?
No, but having a reference genome or transcript annotation improves mapping, APA site identification, and isoform resolution. In non-model organisms, de novo long-read assembly combined with reference-free transcript annotation is possible, though some analyses (e.g. miRNA-binding prediction) may be more challenging without good annotation.
What are the input sample requirements for TAIL Iso-seq?
Total RNA of high integrity is required (recommended RIN ≥ 7), free of contaminants, with sufficient quantity depending on organism / tissue; lower input is possible with optimized methods. The RNA must include intact poly(A) tails and minimal degradation for best results.
How do you handle PCR bias or truncation of poly(A) tails in the pipeline?
We minimise bias by using long-read sequencing (which avoids fragment assembly), capturing full-length transcripts, retaining raw signal data (.fast5) for tail length estimation, and employing QC steps to filter truncated or degraded RNA. Usage of tools that work on raw signal helps ensure accurate tail estimation.
What is the difference between poly(A) tail analysis and APA (alternative polyadenylation) analysis?
Poly(A) tail analysis focuses on the length, composition, and modifications of the tail itself at the transcript-end. APA analysis addresses where polyadenylation occurs—i.e. mapping poly(A) cleavage sites, the varying 3' untranslated region (3'UTR) lengths among isoforms, and how that affects regulation (e.g., miRNA binding). Both are complementary.
Can TAIL Iso-seq be used for comparative studies across conditions or species?
Yes. Because it provides per-isoform tail length distributions and APA site usage per sample, TAIL Iso-seq supports comparisons across tissues, treatments, or species. Statistical pipelines can test for differential tail length or APA usage between groups.
How long will I get the results (bioinformatics + deliverables)?
Typical timelines depend on sample number, complexity, and sequencing depth. Reporting includes raw + processed sequencing data, tail length distributions, APA mapping, isoform catalogues, and visualizations. (Specific turn-around times will be provided based on scope in the project quote.)
How does cost scale with sample number, depth, or target transcript complexity?
Costs generally increase with the number of samples, required read depth, and complexity (e.g. non-model organism or low expression transcripts). N2 Jenomics Lab Pvt. Ltd. offers scalable pricing; clients can choose from standard or premium analysis levels depending on how much resolution (isoform discovery, tail composition, APA precision) they require.