Nanopore Full Lengh Lncrna Sequencing
Home
> Nanopore Full Lengh Lncrna Sequencing
Nanopore Full Length LncRNA Sequencing Service for Isoform Discovery
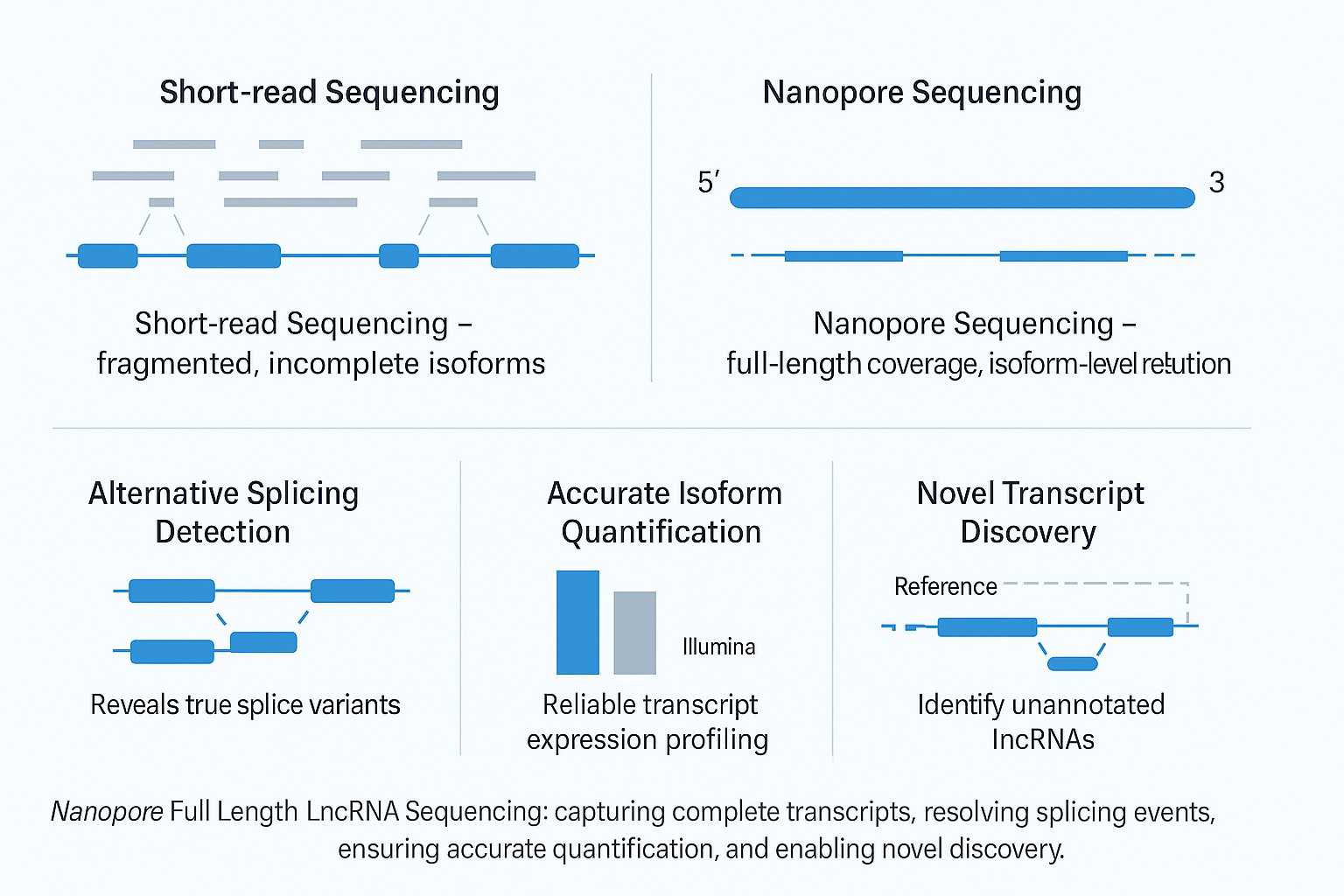
Long non-coding RNAs (lncRNAs) play critical roles in gene regulation, development, and disease, yet most remain poorly characterized. Traditional short-read sequencing fragments transcripts and struggles to reconstruct complete isoforms, limiting discovery and quantification accuracy.
N2 Jenomics Lab Pvt. Ltd. provides a Nanopore Full Length LncRNA Sequencing service that captures full-length transcripts from 5' to 3', enabling isoform-level resolution, splicing detection, and novel lncRNA discovery. Our end-to-end solution empowers researchers with accurate expression profiling and comprehensive transcriptome insights.
Long non-coding RNAs (lncRNAs) are >200 nt transcripts that regulate gene expression, development, epigenetics, and disease pathways. Their functions are often cell-type-specific and highly dependent on alternative splicing. However, short-read sequencing fragments RNA into small pieces, making it difficult to reconstruct complete isoforms or quantify them accurately.
Nanopore Full Length LncRNA Sequencing solves this problem by generating reads that span the entire transcript from 5' to 3'. This enables researchers to identify novel lncRNAs, resolve splice junctions, and quantify isoforms directly—providing a more complete view of the transcriptome.
Our Nanopore Full Length LncRNA Sequencing Solution
N2 Jenomics Lab Pvt. Ltd. ' platform integrates advanced Oxford Nanopore long-read technology with optimized RNA library preparation to deliver accurate, isoform-level insights into the lncRNA transcriptome.
Technical Parameters
Input: Total RNA (≥2 µg, RIN ≥7, rRNA-depleted)
Read length: up to 10–20 kb continuous single-molecule reads
Coverage: single-transcript 5' to 3' sequencing without fragmentation
Throughput: millions of reads per run, scalable for large transcriptomes
Data outputs: FASTQ, BAM/GTF alignment, isoform annotations, quantification matrices
Technical Advantages
Complete transcript coverage – direct sequencing of entire lncRNAs, eliminating the need for assembly
Splicing resolution – reliable identification of exon skipping, intron retention, and complex splice variants
Isoform-level quantification – more accurate measurement of expression compared with short-read approaches
Broad RNA detection – capture of lncRNA, mRNA, tRNA, and small RNA within the same dataset
Poly(A)-independent detection – enables discovery of non-polyadenylated lncRNAs often missed by conventional methods
Short-Read vs. Full-Length LncRNA Library Construction
A clear comparison of short-read RNA-seq and Nanopore full-length lncRNA sequencing highlights why long-read technology delivers more reliable transcriptome insights.
Feature
Short-Read LncRNA Library Construction
Nanopore Full-Length LncRNA Library Construction
RNA Processing
Total RNA → rRNA removal or poly(A) enrichment → RNA fragmentation
Total RNA → rRNA removal → RNA integrity preserved, no fragmentation
Library Preparation
Random priming, cDNA synthesis, multiple PCR cycles, adapter ligation
Full-length cDNA synthesis, minimal amplification, adapter ligation for Nanopore
Read Length & Coverage
50–300 bp fragments; requires computational assembly
10–20 kb continuous reads spanning transcripts from 5' to 3'
Isoform & Splicing Detection
Predicted by assembly; many events remain unresolved
Direct detection of true splice junctions, exon skipping, and isoform diversity
Quantification Accuracy
Fragment counts prone to bias, especially for long or low-abundance transcripts
Isoform-level quantification with consistent, reproducible expression profiles
Novel Transcript Discovery
Limited to known annotations and poly(A)+ lncRNAs
Detects unannotated and non-poly(A) lncRNAs with broad RNA coverage
Bias & Error Sources
Fragmentation and PCR introduce assembly errors, 3'/5' bias
Higher raw error rate, but reduced assembly bias; corrected via bioinformatics
Traditional short-read sequencing is powerful for gene-level expression studies but struggles with isoform reconstruction and alternative splicing detection. In contrast, Nanopore full-length lncRNA sequencing provides continuous read coverage across entire transcripts, enabling accurate isoform quantification, novel lncRNA discovery, and mechanistic insights into transcriptome regulation.
Applications in Research
Nanopore Full Length LncRNA Sequencing supports a wide range of biological and biomedical studies:
Developmental Biology – build lncRNA atlases across tissues, species, and stages of differentiation
Oncology – uncover isoform-level biomarkers and splicing regulation in cancer progression
Neuroscience & Immunology – analyze lncRNA regulation in immune response and neural processes
Environmental Toxicology – track lncRNA expression changes under chemical or pollutant exposure
Evolutionary Biology – identify conserved motifs, sORFs, and miRNA-binding sites in novel lncRNAs
Service Workflow
From consultation to results, N2 Jenomics Lab Pvt. Ltd. delivers a complete solution:
Project consultation – design tailored to your research goals
Sample QC – rRNA depletion and RNA quality control
Library preparation – full-length cDNA synthesis and adapter ligation
Nanopore sequencing – long-read coverage of lncRNA isoforms
Our standard analysis workflow includes several key steps: quality control, alignment, assembly, filtering, quantification, differential lncRNA analysis, differential mRNA analysis, lncRNA target mRNA prediction, and functional enrichment. At the core of this analysis is the assessment of gene expression differences for statistical significance. We compare gene expression across two or more conditions (e.g., treatment vs. control), identify differentially expressed genes associated with specific conditions, and further investigate the biological significance of these genes.
lncRNA-mRNA Association Analysis
LncRNAs regulate target gene expression through co-location or co-expression. For lncRNA-mRNA association analysis, N2 Jenomics Lab Pvt. Ltd. employs an intersection analysis approach: comparing the target genes of differentially expressed lncRNAs with differentially expressed mRNAs. When a target gene of a differentially expressed lncRNA is also a significantly differentially expressed mRNA, it is more likely to be directly or indirectly regulated by the lncRNA.
LncRNA-miRNA Association Analysis
LncRNAs can contain miRNA binding sites, allowing miRNAs to bind to lncRNAs and influence their function. According to the competitive endogenous RNA (ceRNA) theory, miRNAs can bind to lncRNAs through the RNA-induced silencing complex (RISC), leading to lncRNA degradation. To predict target lncRNAs for miRNAs, bioinformatic software is used to identify lncRNAs targeted by differentially expressed miRNAs.
LncRNA, miRNA, and mRNA Association Analysis
LncRNAs possess miRNA binding sites and can competitively bind miRNAs with mRNAs, inhibiting miRNA-mediated regulation of target genes. This indirectly regulates gene expression. Based on the ceRNA theory, we identify lncRNA-target gene pairs that share identical miRNA binding sites. This allows us to construct lncRNA-miRNA-mRNA regulatory networks, where lncRNAs act as decoys, miRNAs as the core, and mRNAs as the targets.
Why Partner with N2 Jenomics Lab Pvt. Ltd.
Over a decade of expertise in next-generation and long-read sequencing
Proven track record with Oxford Nanopore technologies
Dedicated bioinformatics team specializing in transcriptome analysis
Flexible, research-use-only solutions for academia, biotech, and pharma
Reliable data quality, CRO-grade standards, and secure data delivery
Deliverables
You will receive:
Raw data files (FASTQ)
Processed alignment files (BAM/GTF)
Isoform annotation and expression quantification tables
Comprehensive analysis report (PDF + Excel)
Visualizations of isoform structures and splicing events
Nanopore captures complete isoforms and resolves splicing events, avoiding assembly artifacts from fragmented reads.
Q2. Can non-poly(A) lncRNAs be detected?
Yes. Our protocol does not rely on poly(A) enrichment, allowing for broader transcript coverage.
Q3. What sequencing depth is recommended?
We recommend 10–20 million reads for standard transcriptome profiling, with higher depth for low-abundance lncRNAs.
Q4. Do you provide bioinformatics support?
Yes. We deliver isoform-level analysis, quantification, functional annotation, and custom pipelines tailored to your study.
Q5. What other Nanopore services do you provide?
We also offer Nanopore Target Sequencing and multi-transcript profiling options.
Demo Results
Integrated Demo Results of Nanopore Full Length LncRNA Sequencing
Nanopore Full Lengh Lncrna Seq FAQs
What is "full-length lncRNA sequencing" and how does it differ from standard RNA-seq?
Full-length lncRNA sequencing refers to sequencing strategies (typically via long-read technologies) designed to capture transcripts from the 5' end through splice junctions to the 3' end, so you get the full isoform without relying heavily on assembly of short fragments. Standard (short-read) RNA-seq often requires breaking RNA into pieces, assembles reads computationally, which can miss splice variants, exon boundaries, transcription start or end, and low-abundance transcripts. Full-length sequencing improves detection of novel isoforms, accurate splice-junction mapping, and quantification of complete transcripts.
Do you need special sample quality for Nanopore full-length lncRNA sequencing?
Yes, sample quality has a strong impact: total RNA must be of high integrity (low degradation), with good purity (low protein, phenol, or salt contamination), and efficient removal of rRNA. Because full-length reads are more sensitive to RNA breaks, starting with intact RNA improves yield of full transcripts. Low-abundance and long transcripts are especially vulnerable to degradation.
Can we detect novel lncRNAs or splice variants that are not in the reference annotation?
Absolutely. One of the major strengths of full-length lncRNA sequencing is its ability to uncover unannotated transcripts, previously unrecognized splice junctions, and novel isoforms. Because reads cover exon structure completely (including alternative splice sites), you can identify variants that short-read methods often miss.
How accurate is expression quantification with full-length lncRNA sequencing compared to short-read?
Expression quantification is more reliable at the isoform level when you have full-length reads, because you minimize bias from assembly, fragment length, and fragmentation. While base-level error rates may be somewhat higher in long-read technologies, downstream correction and alignment tools allow very good quantification, especially for moderate to high expression transcripts.
Which RNA types can be captured in this sequencing? Does it have to be poly(A)-tailed lncRNAs?
You can capture a broad spectrum: poly(A) lncRNAs, non-poly(A) lncRNAs, mRNAs, and other long non-coding transcripts depending on library prep protocol (e.g. rRNA depletion instead of poly(A) enrichment). Non-poly(A) lncRNAs often require rRNA removal + custom priming or adaptor ligation methods.
What are typical read lengths and output quality for this service?
Typical full-length reads span entire transcript lengths (several kb long), depending on transcript length and quality of input RNA. Quality includes consistent splice junction detection, coverage from 5' to 3' ends, low fragmentation bias. Output includes raw reads, aligned isoforms, and reproducible expression estimates.
Is this type of sequencing suitable for discovering novel biomarkers or in translational / clinical research?
Yes. Because full-length lncRNA sequencing resolves entire isoform structure and enables detection of previously unannotated transcripts or splice variants, it is highly valuable for biomarker discovery, disease mechanism studies, and translational research. With proper sample handling, validation, and bioinformatics pipelines, results can support downstream assays or diagnostic marker validation.
Nanopore Full Lengh Lncrna Seq Case Studies
Reference: Yu J, Qiu K, Sun W, et al. A long noncoding RNA functions in high-light-induced anthocyanin accumulation in apple by activating ethylene synthesis. Plant Physiology. 2022;189:66–83. https://doi.org/10.1093/plphys/kiac049
1. Background
Anthocyanin pigments determine the red coloration of apple peels, directly influencing fruit quality and consumer preference. While light and ethylene are known regulators, the molecular mechanisms linking these signals remained unclear. The study investigated whether long noncoding RNAs (lncRNAs) contribute to the regulation of anthocyanin biosynthesis during high-light exposure in apple (Malus domestica).
2. Methods
Transcriptome sequencing of apple peels exposed to different light conditions was performed to identify light-responsive lncRNAs.
Weighted gene co-expression network analysis (WGCNA) identified MdLNC610 as highly correlated with anthocyanin and ethylene production.
Functional validation included:
RT-qPCR expression analysis under light and inhibitor treatments.
Overexpression and silencing of MdLNC610 and MdACO1 in apple fruit and calli using Agrobacterium-mediated transformation.
Hi-C chromatin conformation capture to assess physical proximity between MdLNC610 and MdACO1.
3. Results
High-light treatment significantly increased both anthocyanin accumulation and ethylene production in apple fruit.
MdLNC610 expression was induced by high light and positively correlated with MdACO1, a key ethylene biosynthesis gene.
Overexpression of MdLNC610 led to higher ethylene levels and enhanced red coloration, while silencing suppressed pigmentation.
Hi-C data confirmed physical association between MdLNC610 and MdACO1, suggesting a cis- or trans-regulatory mechanism.
Functional validation of lncRNA MdLNC610: Overexpression promotes ethylene production and anthocyanin accumulation, while silencing suppresses coloration in apple fruit under high-light conditions.
4. Conclusions
This case demonstrates that Nanopore full-length lncRNA sequencing and integrative transcriptomics can identify novel regulatory lncRNAs like MdLNC610, which mediate key physiological traits such as apple peel coloration. By capturing complete isoforms and resolving expression patterns, long-read sequencing provides critical insights into noncoding RNA–mRNA regulatory networks.
References:
Kyle Palos, et al. Identification and functional annotation of long intergenic non-coding RNAs in Brassicaceae. Plant Cell, 2022.
Li N, et al. A novel trans-acting lncRNA of ACTG1 that induces the remodeling of ovarian follicles. International Journal of Biological Macromolecules. 2023
Yu J, Qiu K, Sun W, et al. A long non-coding RNA functions in high-light-induced anthocyanin accumulation in apple by activating ethylene synthesis. Plant Physiology, 2022.
Lan Z, et al. The interaction between lncrna snhg6 and hnrnpa1 contributes to the growth of colorectal cancer by enhancing aerobic glycolysis through the regulation of alternative splicing of PKM. Frontiers in Oncology, 2020.
Cao M X,et al. Identification of potential long noncoding RNA biomarker of mercury compounds in zebrafish embryos. Chemical Research in Toxicology, 2019.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India