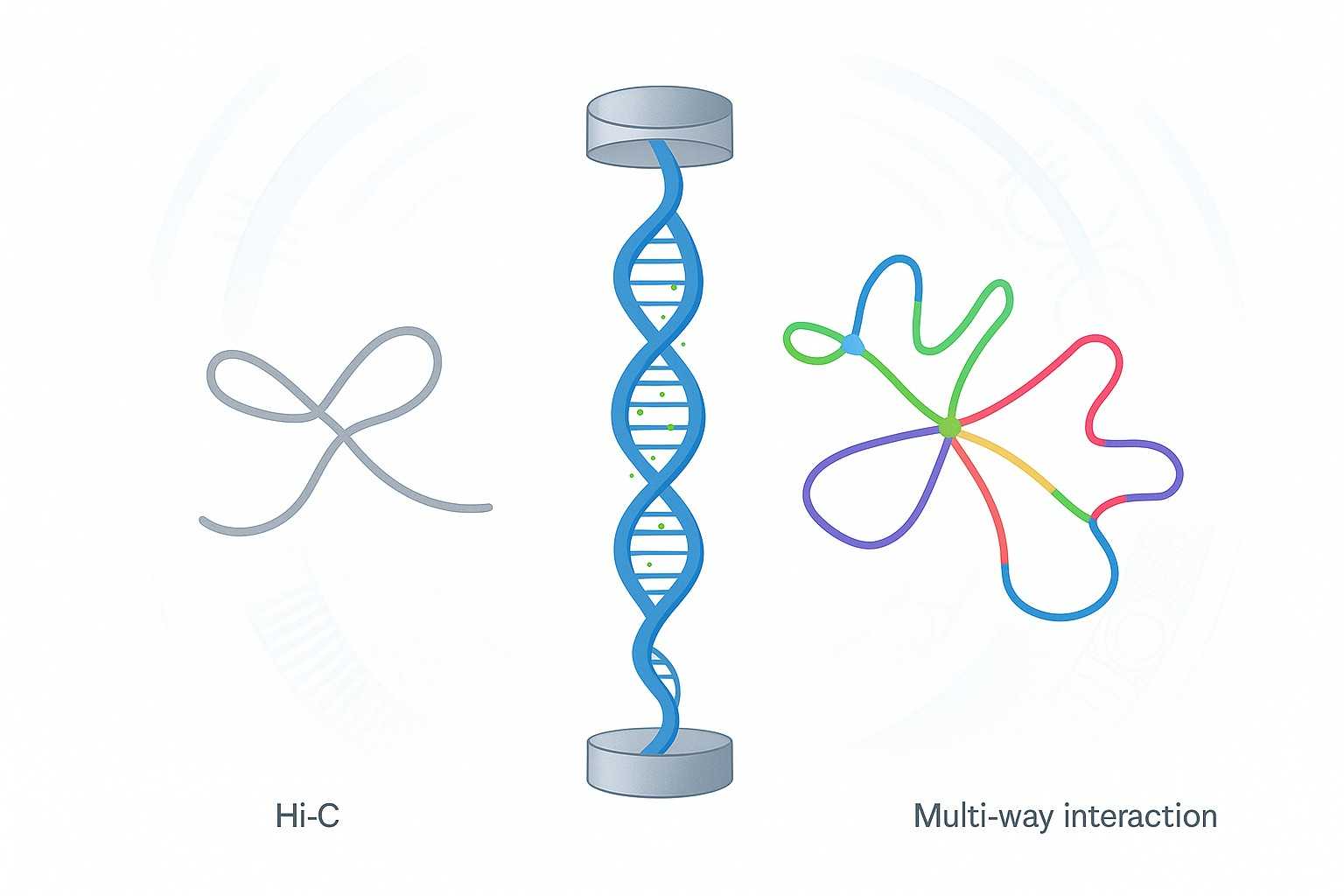
Pore-C Sequencing: Multi-Way Chromatin Mapping With Methylation
N2 Jenomics Lab Pvt. Ltd. provides Pore-C sequencing, a Nanopore-based chromatin conformation capture method that directly reveals multi-way chromatin interactions and DNA methylation in a single experiment. Unlike traditional Hi-C, Pore-C leverages ultra-long Nanopore reads to generate more accurate scaffolding and deeper insights into 3D genome organization.
Our end-to-end service supports biochemistry labs, CRO clients, and academic institutions seeking advanced solutions for telomere-to-telomere assemblies, polyploid genomes, and epigenetic research. From experimental design to pore-c analysis, we deliver high-quality data and actionable results.
Traditional Hi-C sequencing has been the cornerstone for studying 3D genome architecture, but its limitation to pairwise interactions leaves many higher-order chromatin structures unresolved. As genome projects advance to telomere-to-telomere (T2T) assemblies, polyploid organisms, and complex plant and animal genomes, researchers face a growing challenge: How can we accurately anchor contigs, resolve centromeres, and capture true chromatin complexity?
Pore-C sequencing, a long-read Nanopore-based chromatin conformation capture method, provides the solution. By directly sequencing ultra-long concatemers, Pore-C nanopore technology simultaneously reveals multi-way chromatin interactions and DNA methylation, offering insights beyond the reach of Hi-C. For researchers in biochemistry labs, CRO collaborations, and academic institutions, Pore-C opens a new dimension in genome assembly and epigenetic discovery.
Technology Overview: What is Pore-C Sequencing?
Pore-C sequencing is an advanced Nanopore-based chromatin conformation capture technology that overcomes the limitations of Hi-C. By combining chromatin crosslinking and ligation with Oxford Nanopore long-read sequencing, Pore-C enables direct detection of multi-way chromatin interactions along single DNA molecules.
How It Works
Crosslinking & Digestion: Chromatin interactions are preserved through formaldehyde fixation and restriction enzyme cutting.
Ligation: DNA fragments in close spatial proximity are ligated into concatemers, retaining their 3D contact information.
Nanopore Sequencing: Ultra-long concatemers are sequenced directly, capturing multiple interacting loci within a single read.
Integrated Data: In addition to 3D chromatin interactions, Pore-C sequencing preserves native DNA methylation signals, providing both structural and epigenetic insights in one workflow.
Why It Matters
Unlike Hi-C, which is restricted to pairwise interactions, Pore-C nanopore sequencing delivers higher-order contact maps that reflect the true complexity of chromatin folding.
Multi-way interaction data improve the accuracy of telomere-to-telomere genome assembly, centromere resolution, and polyploid scaffolding.
Built-in methylation detection adds a new dimension for epigenetic research.
Pore-C vs Hi-C: A New Dimension in Genome Analysis
Stronger interaction signals across entire genomes, including centromeres.
More complete genome assemblies in both diploid and polyploid species.
Dual insights: 3D chromatin structure and epigenetic modifications.
Efficient sequencing depth: 30× Pore-C can match 100× Hi-C anchoring power.
Service Workflow
Applications of Pore-C Sequencing
Pore-C sequencing provides a powerful solution for research teams seeking to go beyond the limitations of Hi-C. With the ability to capture multi-way chromatin interactions and detect DNA methylation simultaneously, pore-c nanopore analysis supports a broad spectrum of applications across life sciences and agriculture.
Genome Assembly and Scaffolding
Strengthens contig anchoring in telomere-to-telomere (T2T) genome projects.
Improves resolution of centromeres, repetitive regions, and structural variants.
Delivers superior scaffolding accuracy for complex or large genomes.
Polyploid Genome Research
Reduces misassemblies by minimizing homologous chromosome cross-signals.
Provides clearer separation of subgenomes in polyploid plants and animals.
Supports accurate haplotype phasing and evolutionary studies.
3D Genome Architecture
Enables mapping of higher-order chromatin structures beyond pairwise contacts.
Reveals multi-way interaction networks that govern gene regulation.
Advances fundamental understanding of nuclear organization.
Epigenetics and Chromatin Biology
Detects DNA methylation patterns along with structural interactions.
Offers an integrated view of epigenetic regulation in 3D space.
Facilitates research into gene expression control, imprinting, and silencing.
Plant and Animal Breeding
Accelerates pan-genome construction and diversity studies.
Supports identification of structural features linked to agronomic and disease-resistance traits.
Provides high-value insights for genetic improvement programs.
Recommended Pore-C Strategies for Different Genome Levels
To maximize the benefits of Pore-C sequencing in genome assembly, different strategies are recommended depending on the target genome level. These combinations ensure optimal balance between read accuracy, long-range interactions, and sequencing depth.
The true value of Pore-C sequencing lies not only in generating long-read concatemer data but also in extracting accurate and efficient interaction signals. At N2 Jenomics Lab Pvt. Ltd. , we employ optimized pore-c analysis pipelines to ensure high-quality results for genome assembly and 3D genome research.
Optimized Analysis Strategy
Traditional Hi-C pipelines rely on a cut-first, align-later method, which often underestimates the unique power of Pore-C. Instead, we use an align-first, cut-later approach tailored for Nanopore data:
Full-read alignment to the reference genome preserves long-range context.
Post-alignment fragment parsing avoids over-splitting and reduces multi-mapping errors.
Accurate valid pairs extraction increases usable data rates and interaction resolution.
This strategy significantly improves the valid data rate compared with conventional methods, ensuring that each Pore-C read contributes more effectively to downstream analysis.
New QC Metrics for Pore-C
Beyond standard contact map generation, our bioinformatics workflow evaluates Pore-C datasets with specialized metrics, including:
Mean Fragment Count – average number of loci captured per read.
Contacts/Reads Ratio – efficiency of interaction capture per read.
Valid Size/Total Size – proportion of data effectively used.
Mean Valid Pairs Length – distance spanned by valid interactions.
These metrics provide deeper insights into both data quality and biological signal strength, ensuring reliable interpretation.
Deliverables
Clients receive a complete data package that can be directly integrated into genome and epigenetic studies:
Raw Nanopore sequencing data (FASTQ/FAST5)
Processed interaction pairs (multi-way contacts)
Chromatin contact maps and 3D interaction networks
DNA methylation profiles alongside structural data
Comprehensive bioinformatics report with QC statistics and visualizations
Pore-C Data Quality Assurance
Pore-C sequencing offers high-quality data generation with a focus on optimal sample types for reliable results.
The sequencing index
yield (/cell)
Conventional species
pass reads N50≥1Kb
Raw data≥50Gb
Sample description
1. Plant samples are recommended to send newly grown young leaves. 2. Animal samples are recommended to give preference to fresh blood, followed by liver and other tissues. 3. Unconventional samples do not guarantee sequencing yield. 4. Nanopore sequencing unconventional samples are as follows: 1) plants with a lot of secondary metabolites; 2) oceans and aquatic products (animals and plants, algae, etc.); 3) rabbits, insects, amphibians, birds, etc.
Sample Requirements for Pore-C Sequencing
To ensure high-quality results, please follow the guidelines below when preparing and shipping your samples. All samples must be snap-frozen in liquid nitrogen, stored at −80 °C, and shipped on dry ice to avoid degradation.
Sample Type
Recommended Amount
Cell
≥ 106 cells
Peripheral blood mononucleocytes (PBMCs)
PBMC pellet from ~5 ml fresh whole blood
Animal tissue
~50-100 mg cryo-ground tissue
Insect material
~50-100 mg cryo-ground material
C. elegans material
~1 ml cryo-ground worm powder
Plant material
~2 g plant material
Important Notes:
Label each tube clearly with sample ID (letters + numbers) and ensure it matches the Sample Information Form.
Avoid freeze–thaw cycles.
Do not exceed 5× the recommended input amounts, unless otherwise specified for low-yield samples.
From advanced sequencing platforms to high-quality data delivery, N2 Jenomics Lab Pvt. Ltd. offers an efficient, end-to-end solution tailored to diverse research needs. our team ensures reliable results with flexible support.
Proven Expertise: Our team has extensive experience in chromatin conformation techniques, including pioneering work with Pore-C since 2022.
Optimized, Robust Protocols: We have refined every step (crosslinking, lysis, ligation, cleanup) for maximum yield and complexity, minimizing biases.
High-Throughput Sequencing: Direct access to PromethION 48 platforms ensures deep sequencing coverage rapidly.
Advanced Bioinformatics: Beyond standard pipelines, we offer sophisticated multi-way contact analysis, integration, and bespoke solutions.
Rigorous Quality Control: Multiple QC checkpoints guarantee data reliability.
End-to-End Support: From project design consultation to final interpretation support.
Pore-C Seq FAQs
Q: What is Pore-C, and how is it different from Hi-C?
Pore-C is a chromatin conformation capture method that uses long-read Nanopore sequencing instead of short reads; it captures multi-way (3 or more loci) chromatin interactions in single reads, preserves DNA methylation, simplifies library prep by eliminating biotin labeling and PCR steps, and resolves complex regions that are challenging for Hi-C.
Q: Can Pore-C detect epigenetic modifications and interactions at the same time?
Yes, because Pore-C uses Nanopore sequencing which is PCR-free and retains native DNA base modifications; thus both chromatin interaction networks and methylation signatures (for example 5mC) can be obtained from the same dataset.
Q: What types of samples are suitable for Pore-C sequencing?
Samples including crosslinked cells, tissues from plants or animals, fresh or frozen biological material that can preserve chromatin structure are suitable; Pore-C has been used successfully for human cell lines, plant leaf tissues, animal tissue types, often needing appropriate fixation and purification to retain interaction signals.
Q: What are typical data outputs and file formats from Pore-C analysis?
The Pore-C workflow produces raw long-read FASTQ or BAM/concatemer reads, processed contact pairs, multi-way interaction matrices (cooler/.mcool or HiC style), methylation annotation, and QC metrics such as valid contacts per read, fragment count, inter- versus intra-chromosomal contact ratios.
Q: How much sequencing depth is needed for Pore-C to achieve useful genome assembly or interaction mapping?
Required depth depends on genome size and complexity: for chromosome-level scaffolding moderate coverage combined with Pore-C often suffices; for telomere-to-telomere assemblies or polyploid genomes higher long-read and Pore-C coverage improves results; Pore-C generally achieves scaffolding comparable to much higher depth Hi-C with fewer bases when properly processed.
Q: What bioinformatics workflows are used for Pore-C data analysis?
Typical pipelines use an "align-first, then fragment" approach, mapping full long reads to reference genome, then parsing ligation fragments based on restriction enzyme cut sites, extracting multi-contact information and pairwise contacts, generating contact maps, methylation profiling, QC and visualization; tools such as wf-pore-c are commonly used.
Pore-C Seq FAQs
Q: What is Pore-C, and how is it different from Hi-C?
Pore-C is a chromatin conformation capture method that uses long-read Nanopore sequencing instead of short reads; it captures multi-way (3 or more loci) chromatin interactions in single reads, preserves DNA methylation, simplifies library prep by eliminating biotin labeling and PCR steps, and resolves complex regions that are challenging for Hi-C.
Q: Can Pore-C detect epigenetic modifications and interactions at the same time?
Yes, because Pore-C uses Nanopore sequencing which is PCR-free and retains native DNA base modifications; thus both chromatin interaction networks and methylation signatures (for example 5mC) can be obtained from the same dataset.
Q: What types of samples are suitable for Pore-C sequencing?
Samples including crosslinked cells, tissues from plants or animals, fresh or frozen biological material that can preserve chromatin structure are suitable; Pore-C has been used successfully for human cell lines, plant leaf tissues, animal tissue types, often needing appropriate fixation and purification to retain interaction signals.
Q: What are typical data outputs and file formats from Pore-C analysis?
The Pore-C workflow produces raw long-read FASTQ or BAM/concatemer reads, processed contact pairs, multi-way interaction matrices (cooler/.mcool or HiC style), methylation annotation, and QC metrics such as valid contacts per read, fragment count, inter- versus intra-chromosomal contact ratios.
Q: How much sequencing depth is needed for Pore-C to achieve useful genome assembly or interaction mapping?
Required depth depends on genome size and complexity: for chromosome-level scaffolding moderate coverage combined with Pore-C often suffices; for telomere-to-telomere assemblies or polyploid genomes higher long-read and Pore-C coverage improves results; Pore-C generally achieves scaffolding comparable to much higher depth Hi-C with fewer bases when properly processed.
Q: What bioinformatics workflows are used for Pore-C data analysis?
Typical pipelines use an "align-first, then fragment" approach, mapping full long reads to reference genome, then parsing ligation fragments based on restriction enzyme cut sites, extracting multi-contact information and pairwise contacts, generating contact maps, methylation profiling, QC and visualization; tools such as wf-pore-c are commonly used.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India