Nanopore Full-Length Transcripts Sequencing
Home
> Nanopore Full-Length Transcripts Sequencing
Nanopore Full-Length Transcriptome Sequencing Service
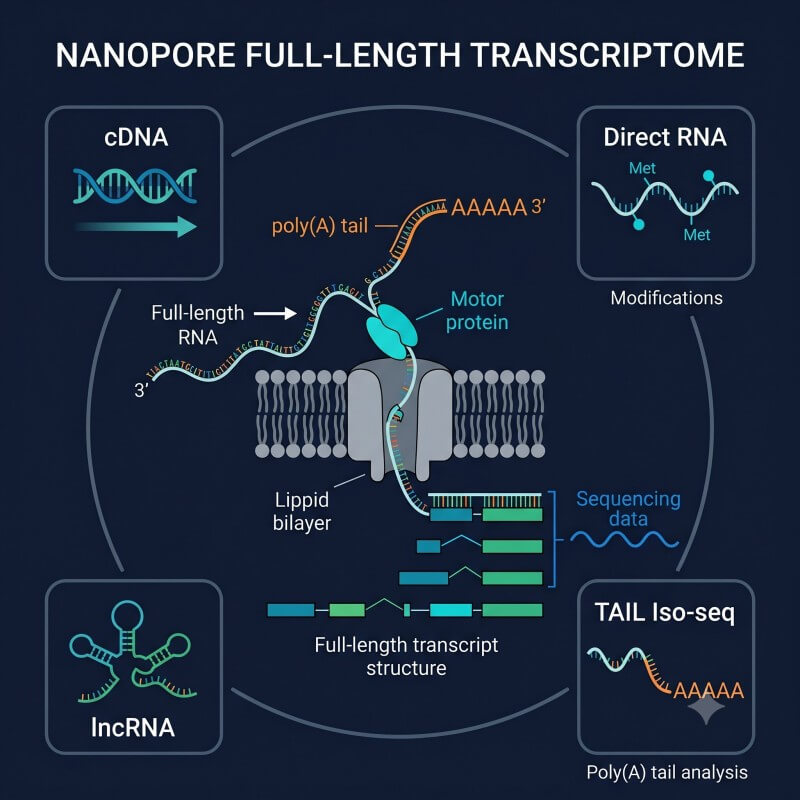
N2 Jenomics Lab Pvt. Ltd. offers comprehensive Nanopore full-length transcriptome sequencing services covering cDNA, lncRNA, Direct RNA, and TAIL Iso-seq approaches. From standard isoform profiling to native RNA modification detection and poly(A) tail analysis — all from a single Oxford Nanopore platform.
Four complementary ONT RNA sequencing approaches in one service
Full-length isoform discovery from 5′ end to poly(A) tail
Direct RNA sequencing with native modification detection (m6A, m5C, Ψ)
TAIL Iso-seq for poly(A) tail length and APA analysis
Dedicated lncRNA sequencing for non-poly(A) long non-coding RNAs
End-to-end service: sample to publication-ready data and analysis
What Is Nanopore Full-Length Transcriptome Sequencing?
Full-length transcriptome sequencing uses Oxford Nanopore long-read technology to sequence complete RNA molecules from 5′ end to 3′ poly(A) tail — without fragmentation. Unlike short-read RNA-seq, which reconstructs transcripts computationally from fragmented reads, Nanopore full-length transcriptome sequencing captures each transcript as a single contiguous read, enabling isoform-level resolution that short-read methods cannot achieve.
A 2025 systematic benchmark by the SG-NEx consortium demonstrated that Nanopore long reads detected 32.7% more exon-skipping events than short-read RNA-seq and identified 1,531 novel multi-exon transcripts and 106 high-confidence fusion genes across seven human cell lines (Chen et al., Nature Methods, 2025). The study also found that long-read data achieved substantially higher accuracy for transcript-level quantification compared to short-read methods (Spearman r = 0.93 vs. 0.49 against spike-in controls).
N2 Jenomics Lab Pvt. Ltd. provides four complementary Nanopore full-length transcriptome sequencing approaches — cDNA, Direct RNA, lncRNA, and TAIL Iso-seq — allowing researchers to choose the method best suited to their biological question.
Why Choose Nanopore for Full-Length Transcriptome Sequencing?
Nanopore full-length transcriptome sequencing offers three distinct advantages that other platforms cannot match.
1
Full-length isoform resolution
Nanopore reads span entire transcripts, allowing direct detection of alternative splicing, fusion genes, and novel isoforms without computational inference.
2
Direct RNA sequencing
ONT is the only commercially available platform that sequences native RNA molecules directly, preserving base modifications (m6A, m5C, pseudouridine) that are lost during reverse transcription.
3
Flexible service portfolio
Four complementary approaches under a single service umbrella, each optimized for different research questions — from standard isoform profiling to lncRNA discovery and poly(A) tail analysis.
N2 Jenomics Lab Pvt. Ltd. offers four distinct Nanopore RNA sequencing approaches, each optimized for different research questions.
Nanopore Full-Length cDNA Sequencing
Full-length cDNA sequencing uses reverse transcription to generate cDNA from poly(A)-enriched RNA, followed by Nanopore sequencing. This approach delivers the highest throughput for isoform discovery, gene expression quantification, and alternative splicing analysis.
Best for: Transcriptome annotation, differential isoform expression, novel transcript discovery, alternative splicing analysis.
Nanopore Full Length LncRNA Sequencing
Targeted full-length lncRNA sequencing captures both poly(A) and non-poly(A) long non-coding RNA molecules, providing complete isoform structures that short-read methods cannot resolve due to low expression levels and repetitive regions common in lncRNAs.
Best for: lncRNA isoform discovery, lncRNA functional annotation, non-poly(A) RNA profiling, eRNA and circRNA identification.
Nanopore Direct RNA Sequencing
Direct RNA sequencing sequences native RNA molecules without reverse transcription or PCR amplification. It preserves all base modifications (m6A, m5C, pseudouridine, inosine) and provides simultaneous isoform and epitranscriptomic information from the same read. Learn more
TAIL Iso-seq combines full-length transcript sequencing with intact poly(A) tail preservation, enabling per-molecule measurement of poly(A) tail length, alternative polyadenylation (APA) site usage, and non-A residue detection — all at isoform resolution. Learn more
Best for: Poly(A) tail length analysis, APA mapping, mRNA stability studies, translational regulation research.
Capability Comparison Across Transcriptome Approaches
Analysis
NGS RNA-seq
PacBio Iso-Seq
ONT cDNA
TAIL Iso-seq
Full-Length lncRNA
Direct RNA
Gene quantification
+++
+
+++
+++
+++
+++
Differential gene expression
+++
+
+++
+++
+++
+++
Isoform quantification
+
++
+++
+++
+++
+++
Differential isoform expression
+
++
+++
+++
+++
+++
Alternative splicing
+
+++
+++
+++
+++
+++
Fusion gene detection
+
+++
+++
+++
+++
+++
Novel transcript prediction
+
+++
+++
+++
+++
+++
Allele-specific expression
—
+++
+++
+++
+++
+++
Alternative polyadenylation (APA)
+
+++
+++
+++
+++
+++
Poly(A) tail length
—
—
—
+++
—
+++
lncRNA analysis
+
+
+
+
+++
+
RNA modifications
—
—
—
—
—
+++
Strand-specific analysis
—
+++
+++
+++
+++
+++
+++ = Optimal / Gold Standard | ++ = Good | + = Basic capability | — = Not applicable
Technology and Workflow
Our Nanopore full-length transcriptome sequencing workflow is designed for reproducibility and quality at every stage.
1. Sample QC
RNA integrity assessed by RIN score (Agilent Bioanalyzer), quantity by Qubit fluorometer, purity by spectrophotometry (OD260/280, OD260/230). QC pass threshold varies by approach.
Library quality assessed by fragment size distribution and concentration. Minimum pass: appropriate size profile without adapter dimers or short fragment contamination.
4. Nanopore Sequencing
Performed on PromethION or GridION platforms with R10.4.1 flow cells and Dorado basecalling. Real-time data monitoring allows informed decisions on run duration.
5. Basecalling and Demultiplexing
Dorado super-accurate basecalling model with quality filtering. Barcode demultiplexing with zero mismatches allowed. Modified base detection enabled for Direct RNA data.
6. Data Delivery
Raw FASTQ, full-length read consensus sequences, and comprehensive QC report delivered via secure download or physical drive.
Sample Requirements
Proper RNA quality is essential for successful full-length transcriptome sequencing. Below are the general recommendations for each approach.
Sample requirements may be adjusted based on project-specific conditions. Contact our team for a detailed consultation before sample submission.
Shipping guidelines: Ship total RNA on dry ice in RNase-free tubes. For Direct RNA projects, flash-frozen tissue in liquid nitrogen is preferred over RNA stabilization reagents.
Bioinformatics Analysis
Our bioinformatics pipeline is designed to deliver actionable transcriptome data, not just raw sequences.
Transcript identification — Alignment to reference genome (minimap2) or reference-free clustering (isONform); full-length non-chimeric (FLNC) read generation
Isoform quantification — Gene-level and isoform-level expression matrices (TPM / raw counts)
Alternative splicing analysis — Detection and classification of exon skipping, intron retention, alternative 5′/3′ splice sites, mutually exclusive exons
lncRNA identification and classification (CPC2, CNCI, Pfam); target gene prediction
Fusion gene detection and validation from chimeric reads
APA mapping and poly(A) tail length distribution analysis
RNA modification detection (m6A, m5C, pseudouridine) from Direct RNA data
Novel transcript annotation with evidence-based transcript model generation
Deliverables
N2 Jenomics Lab Pvt. Ltd. provides comprehensive and organized deliverables for every full-length transcriptome project, tailored for seamless downstream analysis and publication.
Deliverable
Description
Raw sequencing data
FASTQ files per sample (Dorado basecalled, demultiplexed)
Full-length reads
FLNC consensus sequences in FASTA format
Transcript quantification
Gene-level and isoform-level expression matrices (TPM / counts)
Isoform annotation
GTF file with transcript models and exon structures
Alternative splicing report
Classification and quantification of AS events per sample
Differential analysis
DEG and DET results with statistical testing
QC report
Comprehensive quality metrics for each sample and run
Nanopore full-length transcriptome sequencing supports a broad range of research applications across transcriptomics and epitranscriptomics.
Transcriptome Annotation and Isoform Discovery — Nanopore full-length sequencing provides direct evidence for transcript models, making it the method of choice for genome annotation projects. The SG-NEx benchmark identified over 1,500 novel multi-exon transcripts and refined existing gene models across multiple human cell lines (Chen et al., 2025).
Alternative Splicing and Disease Research — Long-read RNA sequencing captures complete splice variants in single reads, enabling reliable detection of disease-associated splicing events that short-read methods often miss (Santucci et al., 2024).
Epitranscriptomics and RNA Modification Mapping — Direct RNA sequencing detects m6A, m5C, pseudouridine, and inosine at single-base resolution without antibody enrichment, providing simultaneous transcript identity and modification status (Sun et al., 2025).
Cancer Transcriptomics — Full-length transcriptome sequencing enables direct detection of gene fusions, aberrant splicing isoforms, and allele-specific expression — key drivers of tumorigenesis that are challenging to resolve with short-read approaches (Deng et al., 2024).
lncRNA Characterization — Full-length lncRNA sequencing captures complete lncRNA isoforms, including those with repetitive regions or low expression levels that short-read RNA-seq cannot reliably assemble.
Poly(A) Tail and mRNA Stability Studies — TAIL Iso-seq provides isoform-specific poly(A) tail length distributions, enabling researchers to link tail dynamics to translational regulation, mRNA decay, and cellular stress responses.
Comparison: Choosing the Right Transcriptome Solution
When selecting a transcriptome sequencing approach, the choice depends on your research objectives. Use the guidance below to identify the best fit for your project.
Choose NGS RNA-seq when — Your primary goal is gene-level expression quantification at the lowest cost per sample, and isoform-level resolution is not required.
Choose PacBio Iso-Seq when — You need the highest per-read accuracy for novel isoform discovery and can accept lower throughput and higher cost per sample.
Choose Nanopore cDNA when — You need high-throughput full-length isoform profiling with the flexibility to add optional analyses (splicing, fusion genes, APA) at a competitive price point.
Choose Nanopore Direct RNA when — Your research requires native RNA modification detection, minimal technical bias, or simultaneous isoform and epitranscriptomic analysis.
Choose TAIL Iso-seq when — Poly(A) tail length distribution and APA profiling are central to your research question.
Choose Full-Length lncRNA when — Your focus is on long non-coding RNA biology, including non-poly(A) transcripts that standard poly(A)-enriched methods cannot capture.
For a detailed comparison of analytical capabilities across all methods, see the capability matrix in the services section above.
References
Chen et al. "A systematic benchmark of nanopore long-read RNA sequencing for transcript-level analysis in human cell lines." Nature Methods, 2025. https://doi.org/10.1038/s41592-025-02623-4
Kabza et al. "Accurate long-read transcript discovery and quantification at single-cell, pseudo-bulk and bulk resolution with Isosceles." Nature Communications, 2024. https://doi.org/10.1038/s41467-024-51584-3
Deng et al. "Systematic evaluation of single-cell RNA-seq analyses performance based on long-read sequencing platforms." Journal of Advanced Research, 2024. https://doi.org/10.1016/j.jare.2024.05.020
Sun et al. "Advances in nanopore direct RNA sequencing and its impact on biological research." Biotechnology Advances, 2025. https://doi.org/10.1016/j.biotechadv.2025.108710
Petri & Sahlin. "isONform: reference-free transcriptome reconstruction from Oxford Nanopore data." Bioinformatics, 2023. https://doi.org/10.1093/bioinformatics/btad264
Santucci et al. "Enhancing novel isoform discovery: leveraging nanopore long-read sequencing and machine learning approaches." Briefings in Functional Genomics, 2024. https://doi.org/10.1093/bfgp/elae031
Zhang et al. "Nanopore sequencing: flourishing in its teenage years." Journal of Genetics and Genomics, 2025. https://doi.org/10.1016/j.jgg.2025.01.004
For research use only. Not intended for clinical diagnosis, treatment, or individual health assessments.
Demo Results
Read length distribution showing full-length transcript capture (N50 typically >10 k
Isoform structure view — novel isoforms detected with full exon-intron annotatio
Alternative splicing events detected, including exon skipping and intron retention (Chen et al., 2025)
RNA modification detection (m6A) from Direct RNA sequencing data
Poly(A) tail length distribution measured by TAIL Iso-seq at isoform resolution
1. What is the difference between Nanopore cDNA and Direct RNA sequencing?
Nanopore cDNA sequencing converts RNA to cDNA via reverse transcription before sequencing, providing higher throughput but losing RNA modification information. Direct RNA sequencing sequences native RNA molecules directly, preserving all base modifications but requiring higher input amounts. The choice depends on whether modification detection or maximum throughput is the priority.
2. Can I detect RNA modifications with this service?
Yes. Our Direct RNA sequencing service can detect m6A, m5C, pseudouridine, and inosine modifications at single-base resolution using trained basecalling models integrated into the Dorado software suite.
3. What is TAIL Iso-seq and when should I use it?
TAIL Iso-seq is a specialized Nanopore sequencing approach that preserves the poly(A) tail during library preparation, enabling per-molecule measurement of poly(A) tail length and alternative polyadenylation site usage. Use it when poly(A) tail dynamics, mRNA stability, or APA regulation is central to your research question.
4. How much RNA do I need to send?
Standard cDNA and lncRNA projects require ≥1 µg total RNA with RIN ≥7.5 (animal) or ≥7.0 (plant). Direct RNA projects require ≥2 µg high-quality RNA. See the Sample Requirements section for detailed specifications by sample type.
5. What bioinformatics analysis is included in the standard package?
The standard package includes basecalling, transcript identification, gene and isoform quantification, differential expression analysis, alternative splicing detection, and functional annotation. Optional add-ons include lncRNA analysis, fusion gene detection, APA profiling, and RNA modification analysis.
6. Can Nanopore full-length transcriptome sequencing detect lncRNAs?
Yes. Our dedicated lncRNA sequencing service captures both poly(A) and non-poly(A) lncRNAs. Standard cDNA sequencing also detects poly(A) lncRNAs, but the lncRNA-specific workflow provides broader coverage of the lncRNA transcriptome, including low-expression and repetitive lncRNAs.
7. How does this service compare to PacBio Iso-Seq?
PacBio Iso-Seq offers higher per-read accuracy (Q30+ via CCS) and is well-suited for high-confidence novel isoform discovery. Nanopore sequencing provides higher throughput, lower cost per sample, and unique capabilities including Direct RNA sequencing and poly(A) tail profiling. Both platforms enable full-length isoform analysis impossible with short-read NGS (Deng et al., 2024).
8. What species do you support?
We accept RNA samples from any species. Standard analysis requires a reference genome for alignment-based transcript identification. Reference-free analysis (de novo transcript clustering) is available for species without an annotated genome.
Nanopore Full-Length Transcriptome Sequencing Case Studies
Published Research Highlight
Accurate long-read transcript discovery and quantification at single-cell, pseudo-bulk and bulk resolution with Isosceles
Single-cell transcriptomics has transformed our understanding of cellular heterogeneity, but standard short-read methods cannot capture full-length isoform information, leaving cell-type-specific splicing largely unexplored.
Methods
Kabza et al. applied Nanopore long-read sequencing to 951 single-cell transcriptomes from the developing mouse brain (embryonic day 18). They developed Isosceles, a computational toolkit that performs reference-based transcript discovery and quantification from long-read data. The study used Nanopore cDNA sequencing on the PromethION platform and compared results against matched Illumina short-read data from the same single-cell cDNA libraries.
Figure 3 from Kabza et al., Nature Communications, 2024. Single-cell isoform discovery in mouse brain.
Results
The analysis identified 44,325 transcript isoforms, with approximately 40% being novel. Coordinated splicing programs were detected within and between neuronal differentiation lineages — information invisible to short-read analysis. Isosceles demonstrated superior sensitivity over existing methods including Bambu, IsoQuant, and ESPRESSO for isoform detection and quantification.
Conclusion
This study demonstrates that Nanopore full-length transcriptome sequencing at single-cell resolution can uncover cell-type-specific isoform diversity entirely missed by short-read approaches, providing a more complete view of transcriptional regulation in complex tissues.
For research use only. Not intended for clinical diagnosis, treatment, or individual health assessments.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India