Absolute Metagenomic Sequencing Service
Home
> Absolute Metagenomic Sequencing Service
Accurate Absolute Metagenomic Sequencing for ARGs and Microbiom
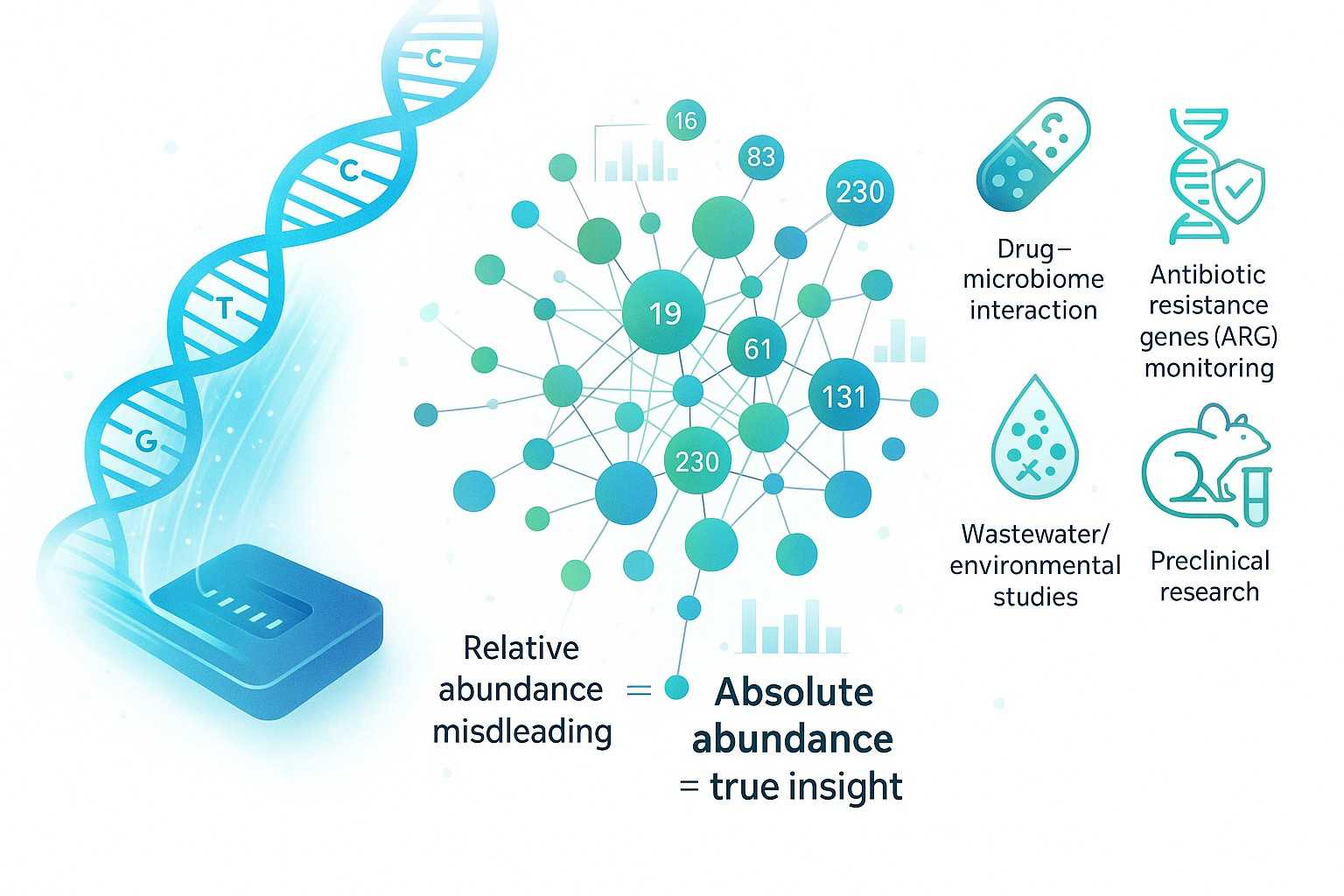
Go beyond relative abundance with absolute metagenomic sequencing powered by Nanopore/PacBio. N2 Jenomics Lab Pvt. Ltd. delivers precise microbial quantification for research labs, CRO projects, and academic institutions.
Key Value:
Unlock absolute abundance microbiome profiling for accurate cross-sample comparison
Achieve strain-level resolution, ARG detection, and host tracking with long-read metagenomics
Preserve critical information on base modifications (6mA, 5mC)
End-to-end solutions from sample prep to bioinformatics, trusted by global researchers
Understanding the microbiome is central to research in human health, drug development, agriculture, and environmental science. Yet, most microbiome studies still rely on relative metagenomic sequencing, which measures proportional composition rather than the true microbial load. This compositional approach can distort microbial dynamics, mask cross-sample differences, and limit biological interpretation.
Our Absolute Metagenomic Sequencing Service overcomes these limitations by combining long-read metagenomics with absolute quantification strategies. By integrating internal standards and spike-in controls, we deliver accurate microbial counts per sample, enabling reliable cross-study comparisons and quantitative risk assessments. This service is designed for academic institutions, biochemistry labs, and CRO projects that require trustworthy microbiome insights beyond relative abundance
Why Absolute Metagenomic Sequencing?
Conventional relative abundance profiling forces microbial communities into percentages that always sum to one. An increase in one species may appear to decrease another, even when both are growing, leading to misleading correlations and false conclusions. For researchers investigating drug–microbiome interactions, environmental microbiology, or antibiotic resistance genes (ARGs), such bias can critically undermine results. Absolute metagenomic sequencing solves this problem by measuring the true abundance of microbial taxa and functional genes. This approach enables:
Accurate microbial load assessment – detect real increases or decreases in taxa without compositional bias.
Cross-sample and cross-study comparability – normalize results to absolute counts, not relative fractions.
Quantitative microbial risk assessment – evaluate pathogens and ARGs in terms of actual copy number per volume.
Enhanced biological interpretation – link microbial shifts to host response, drug treatment, or environmental change with higher confidence.
By integrating absolute abundance microbiome profiling with long-read sequencing, N2 Jenomics Lab Pvt. Ltd. provides both resolution and reliability, helping clients move from relative estimates to true insights.
Comparison: Relative vs. Absolute Abundance
Feature
Relative Abundance
Absolute Abundance
Measure Type
Proportional (% of total)
Actual counts (cells, gene copies per volume or mass)
Dependency
Total community DNA, sequencing depth, compositional effects
Calibrated via spike-in, total microbial load, less impacted by compositional bias
Correlation Risk
High risk of spurious correlations; dominance effects distort interpretations
More direct and interpretable shifts; enables confident detection of real changes
Cross-sample / Cross-study Comparability
Poor, since total read counts or community composition vary widely
Better, because normalized to absolute units allows direct comparison across conditions and studies
Application Scenarios
Good for understanding community structure, diversity, proportions
Better for risk assessment, tracking pathogen load, ARG abundance, drug treatment effects
Platform Comparison & Selection Guide
Platform
Key Strengths
Limitations
Best Use Cases
Nanopore
Ultra-long reads (tens of kb to >1 Mb), real-time data streaming; direct detection of base modifications; excellent for capturing complete genomes, plasmids, mobile elements; portable workflows.
Raw read accuracy lower than "HiFi" PacBio or high-coverage Illumina; more sequencing errors especially in homopolymer regions; may require higher depth or polishing.
When you need ultra-long reads for structural variation, ARG host tracking, rapid/field deployment, or when you want epigenetic modification profiling.
PacBio (HiFi / SMRT)
Combines long reads with high single-molecule accuracy (HiFi), strong for repeat regions, structural variant detection, and high-quality assemblies; lower error rates post-consensus.
Higher cost per base; longer turnaround and instrument cost; may need more input DNA of high quality; lower real-time streaming capability compared to Nanopore.
For projects needing reference-grade genomes, high base accuracy, complex regions, or validation/polishing of long read assemblies; methylation/epigenetics work.
Illumina / Second-Generation Sequencing
Very high per-base accuracy; excellent throughput; lower cost per Gb; well established pipelines; ideal for short fragment sequencing, large numbers of samples.
Short reads make assembly of repeats, plasmids, mobile elements, ARG host linkage difficult; no direct detection of base modifications; relative abundance only unless used with spike-in or microbial load calibration.
When needing high sample throughput, cost sensitivity, comparative studies; or for polishing assemblies from long-read data; for diversity estimation, SNP detection, gene quantification.
How to Choose the Right Platform
If your priority is structural resolution / ARG host linkage / plasmid assembly / epigenetic modifications, lean toward Nanopore or PacBio.
If you need high base accuracy especially for SNPs or rare variant detection, PacBio HiFi or Illumina (or a hybrid approach) may be preferable.
Budget, DNA quantity & quality, and sample type influence choice: long-read platforms often need higher molecular weight DNA.
Often the hybrid strategy (long reads + short reads / polishing) yields the best of both worlds.
Our Absolute Metagenomic Sequencing Workflow
N2 Jenomics Lab Pvt. Ltd. provides an end-to-end absolute metagenomic sequencing workflow powered by long-read technology. Our streamlined process ensures accurate microbial quantification, high-quality assemblies, and actionable results for research clients.
Step 1. Sample Collection & Quality Assessment
Support for diverse sample types: stool, saliva, wastewater, soil, marine water, bioreactor, and extreme environments.
Initial QC to ensure DNA integrity and sufficient microbial load for downstream analysis.
Step 2. DNA Extraction & Spike-In Controls
High-quality microbial DNA extraction with minimal host contamination.
Incorporation of cellular or synthetic spike-in standards to calibrate sequencing data and enable absolute abundance microbiome profiling.
Step 3. Library Preparation & Sequencing
Optimized library preparation tailored to complex metagenomes.
Nanopore/Pacbio sequencing on the latest platforms, generating ultra-long reads for complete microbial genome and plasmid assembly.
Step 4. Data Processing & Assembly
Rigorous quality control of raw reads.
Assembly and binning of metagenome-assembled genomes (MAGs) and plasmids.
Taxonomic classification down to species and strain level.
Annotation of functional genes and metabolic pathways (KEGG, GO, eggNOG).
Detection of antibiotic resistance genes (ARGs) and virulence factors, with host-tracking enabled by long reads.
Optional base modification analysis (6mA, 5mC).
Step 6. Absolute Abundance Analysis & Reporting
Conversion of sequencing data to absolute counts per sample volume using spike-in calibration.
Comprehensive data reports, including relative and absolute abundance tables, functional insights, and customized visualizations.
Delivery of publication-ready results for research, CRO projects, and academic studies.
Applications
Human and animal microbiome research – absolute abundance microbiome profiling for gut, oral, and skin communities
Drug–microbiome interaction studies – evaluate therapeutic effects and safety through absolute metagenomic sequencing
Antibiotic resistance monitoring – ARG detection and host tracking in clinical and environmental samples
Wastewater-based epidemiology (WBE) – rapid surveillance of pathogens and resistance genes in water systems
Environmental microbiology – soil, marine, and extreme environment metagenomics for ecology and biodiversity studies
Industrial microbiology and bioprocess monitoring – track microbial composition and functional genes in production systems
Why N2Jenomics Lab Pvt. Ltd. ?
Choosing the right partner for absolute metagenomic sequencing is critical to obtaining reliable, actionable results. N2 Jenomics Lab Pvt. Ltd. stands out with unique advantages that go beyond standard sequencing providers.
Proven expertise in Long-read metagenomics
Over a decade of experience delivering high-quality Long-read sequencing services, from ultra-long reads to targeted and full-length transcriptome solutions.
Absolute quantification capability
Unlike many competitors that report only relative abundance, we integrate spike-in standards and advanced calibration to provide true microbial load measurements.
Comprehensive deliverables
From absolute abundance microbiome tables to genome assemblies, ARG host tracking, and base modification profiling, we provide a complete view of microbial communities.
Cross-application support
Expertise across human health, drug–microbiome research, environmental monitoring, and industrial microbiology ensures tailored solutions for each client.
End-to-end project management
From sample preparation to advanced bioinformatics and interpretation, we offer one-stop services that save clients time and resources.
Trusted global CRO partner
Serving leading academic institutions, pharmaceutical companies, and biotech firms worldwide with consistent quality and professional support.
N2 Jenomics Lab Pvt. Ltd. delivers not just sequencing data, but actionable microbiome insights—helping our clients move confidently from raw data to meaningful discovery.
Sample Requirements
Sample Type
Recommended Quantity
Minimum Quantity
Concentration
Notes
Genomic DNA
≥ 5 µg
—
≥ 20 ng/µL
High molecular weight DNA, OD260/280 = 1.8–2.0
Long-read Metagenomic DNA
≥ 2 µg
—
≥ 30 ng/µL
DNA should be RNase-free, no degradation/contamination
Environmental Samples
6 g
2 g
—
Soil, sludge, sediment accepted
Water Filter Membrane
6
2
—
0.22 µm membranes recommended for microbial capture
Tissue
2 g
1 g
—
Fresh or frozen, quick-freeze in liquid nitrogen
Interstitial Fluid
6–10 mL
2 mL
—
Store frozen, ship on dry ice
Deliverables
Raw sequencing data with QC report
Taxonomic profiling with relative and absolute abundance tables
High-quality genome and plasmid assemblies (MAGs)
Functional gene and pathway annotation (KEGG, GO, eggNOG)
ARG and virulence factor detection with host tracking
Optional base modification profiling (6mA, 5mC)
Comprehensive project report with publication-ready figures
Demo Results
Absolute Metagenomic Seq FAQs
Q: What is the difference between relative abundance and absolute abundance in metagenomic sequencing?
Absolute abundance refers to measuring the actual number of microbial cells, genes, or taxa per unit sample (e.g. per gram, per mL), which avoids misleading interpretations that arise when data are only expressed as proportions; relative abundance only shows percentages of all detected organisms, so if one taxon increases, another must decrease even if its own absolute level stays constant.
Q: Can equencing provide strain-level resolution and identify antibiotic resistance gene (ARG) hosts?
Yes, long reads enable assembly of metagenome-assembled genomes (MAGs) and plasmids, and they allow ARGs to be mapped to their microbial hosts because long reads span both resistance genes and flanking genomic regions, which helps reveal co-localization and mobile genetic element transfers.
Q: Do I need a lot of DNA to perform absolute metagenomic sequencing?
While high-quality, high molecular weight DNA improves assembly and accuracy, recent studies show that even lower DNA input (tens of nanograms) can yield useful results for community composition and MAG recovery when combined with good sequencing depth and calibration using spike-ins.
Q: How does Absolute Metagenomic Sequencing help in environmental or clinical pathogen monitoring compared to traditional methods?
It offers real-time data streaming, detection of unculturable organisms, direct ARG detection and host tracking, and quantification in absolute terms, which allows earlier detection of potential risks versus culture-based or relative-only sequencing methods which may be slower and less comprehensive.
Q: Can sample types with high host DNA contamination be used for absolute metagenomic sequencing?
They can, but host contamination reduces usable microbial reads, so it's important to reduce host DNA during sample prep or use bioinformatics filtering; even when host background is high, absolute quantification still works with proper spike-in controls and QC to assess usable data fraction.
Q: What kind of bioinformatics and reporting will I receive with this service?
You will receive both relative and absolute abundance tables, taxonomic profiles at strain/metagenome-assembled genomes (MAG) level, functional and pathway annotation, ARG and virulence factor detection with host attribution, QC metrics, assembly statistics, and publication-ready figures to support downstream research or regulatory / CRO deliverables.
Absolute Metagenomic Seq Case Studies
Reference: Zhan J, Cheng J, Chang W, Su Y, Yue X, Wu C. Absolute Quantitative Metagenomic Analysis Provides More Accurate Insights for the Anti-Colitis Effect of Berberine via Modulation of Gut Microbiota. Biomolecules 2025, 15(3):400. https://doi.org/10.3390/biom15030400
Background
Ulcerative colitis (UC) is a chronic inflammatory disease associated with gut microbiota imbalance. Conventional microbiome studies using relative abundance may obscure true microbial dynamics. Berberine (BBR), a natural compound with antimicrobial activity, is reported to modulate gut microbiota, while sodium butyrate (SB) mainly supports beneficial bacterial growth. This study compared relative vs. absolute metagenomic sequencing to evaluate their accuracy in characterizing BBR's anti-colitis effects.
Methods
Animal Model: DSS-induced colitis in mice, divided into control, model, BBR, and SB groups (n=12 each).
Treatments: Oral administration of berberine or sodium butyrate before and during DSS induction.
Sequencing: Both relative quantification and absolute metagenomic sequencing were performed to assess gut microbiota composition and abundance.
Analysis: Microbial richness, diversity, and differential taxa were compared. In addition, a meta-analysis of 13 cohorts was conducted to validate findings.
Results
Symptom Relief: Both BBR and SB improved colitis symptoms, reducing weight loss, colon shortening, and inflammatory cytokine levels.
Relative Quantification Findings: Suggested shifts in microbiota but showed inconsistent results, sometimes opposite to biological reality.
Absolute Quantification Findings: Revealed more accurate bacterial loads. BBR significantly increased beneficial Akkermansia and decreased pathogenic taxa such as Erysipelatoclostridium, aligning with clinical observations.
Meta-Analysis (13 studies): Confirmed that Akkermansia upregulation and Erysipelatoclostridium downregulation were consistent with absolute quantification, but often misrepresented by relative methods.
Figure. Absolute quantitative analysis of gut microbiota after berberine (BBR) and sodium butyrate (SB) treatment in DSS-induced colitis mice. Community richness, diversity, and taxonomic profiles were assessed using absolute abundance data, revealing clearer shifts than relative quantification.
Conclusions
This study demonstrates that absolute quantitative metagenomic sequencing provides a truer representation of microbial community changes than relative quantification. For drug–microbiome studies such as BBR's anti-colitis effect, absolute abundance data:
Avoid spurious correlations,
Capture real microbial shifts,
Offer stronger translational and clinical relevance.
References:
Yang Y, Che Y, Liu L, Wang C, Yin X, Deng Y, Yang C, Zhang T. Rapid absolute quantification of pathogens and ARGs by nanopore sequencing. Sci Total Environ. 2022 Feb 25;809:152190. doi: 10.1016/j.scitotenv.2021.152190. Epub 2021 Dec 7. PMID: 34890655.
Barlow, J.T., Bogatyrev, S.R. & Ismagilov, R.F. A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities. Nat Commun 11, 2590 (2020).
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India