N2 Jenomics Lab Pvt. Ltd. provides Single-Nucleus RNA Sequencing Service for researchers working with frozen, archived, fragile, or difficult-to-dissociate tissues. We help you evaluate sample suitability, generate nuclei-based transcriptomic data, and translate tissue heterogeneity into clustering, marker gene, cell type, and pathway-level insights for research projects.
Profile frozen or difficult tissue samples
Reduce dependence on whole-cell dissociation
Map cell types and cell states from nuclei
Review QC before biological interpretation
Receive analysis-ready files, figures, and reports
Profile Frozen and Difficult Tissue at Nuclear Resolution
Single-nucleus RNA sequencing, often called snRNA-seq, measures gene expression from isolated nuclei rather than intact whole cells. This makes it useful when a high-quality single-cell suspension is hard to obtain, but cell-type-level transcriptomic information is still needed.
For many tissue projects, sequencing is not the first challenge. The real challenge is getting a usable input while preserving the biological signal. Brain, heart, skeletal muscle, adipose-rich tissue, fibrotic tissue, tumor tissue, large cells, and fragile cell populations can be difficult to dissociate into clean whole-cell suspensions. In these situations, nuclei-based profiling can offer a practical route to single-cell-level insight from tissue that may not work well in a standard whole-cell workflow.
We use snRNA-seq to help researchers study cellular heterogeneity, cell-state shifts, rare or underrepresented populations, and sample-group differences in complex tissues. This service is especially relevant when your project involves frozen tissue, limited samples, or tissues where dissociation may introduce stress response, cell loss, or cell-type bias.
When Single-Nucleus RNA Sequencing Is the Better Fit
Single-nucleus RNA sequencing is often a strong fit when your sample cannot reliably produce a clean, viable, single-cell suspension.
The target tissue contains large, fragile, or irregular cells.
Dissociation may change gene expression before capture.
The study needs cell-type-level data from difficult tissue.
You want to compare cell populations across groups or conditions.
snRNA-seq is not automatically the best choice for every project. If fresh, viable cells are available and whole-cell transcriptome capture is important, single-cell RNA sequencing may be a better option. Our team helps you choose the workflow that fits both the sample and the research question.
Research Questions This Service Helps Address
Researchers use single-nucleus RNA sequencing to answer questions such as:
Which cell types are present in this tissue?
Which cell states differ between experimental groups?
Which marker genes define each nucleus cluster?
Are specific cell populations expanded or reduced?
Which pathways are enriched in selected cell types?
How do disease models, treatments, or developmental stages alter tissue composition?
Which cell populations should be prioritized for follow-up assays?
For projects that may need related single-cell services, you can also review our Single-Cell Sequencing platform.
Suitable Study Areas
Single-nucleus RNA sequencing can support a wide range of tissue-focused research programs, including:
Research Area
How snRNA-seq Helps
Neuroscience
Profiles nuclei from brain regions where intact cell recovery can be difficult.
Oncology
Helps resolve tumor, stromal, and immune-related cell states in complex tissue.
Immunology
Supports tissue-level immune microenvironment studies when tissue dissociation is limiting.
Developmental biology
Maps cell-state transitions across tissue stages or experimental conditions.
Organoid research
Compares cellular composition and differentiation states across models.
Drug response studies
Identifies cell-type-specific transcriptomic changes after perturbation.
From Tissue or Nuclei Input to Sequencing-Ready Libraries
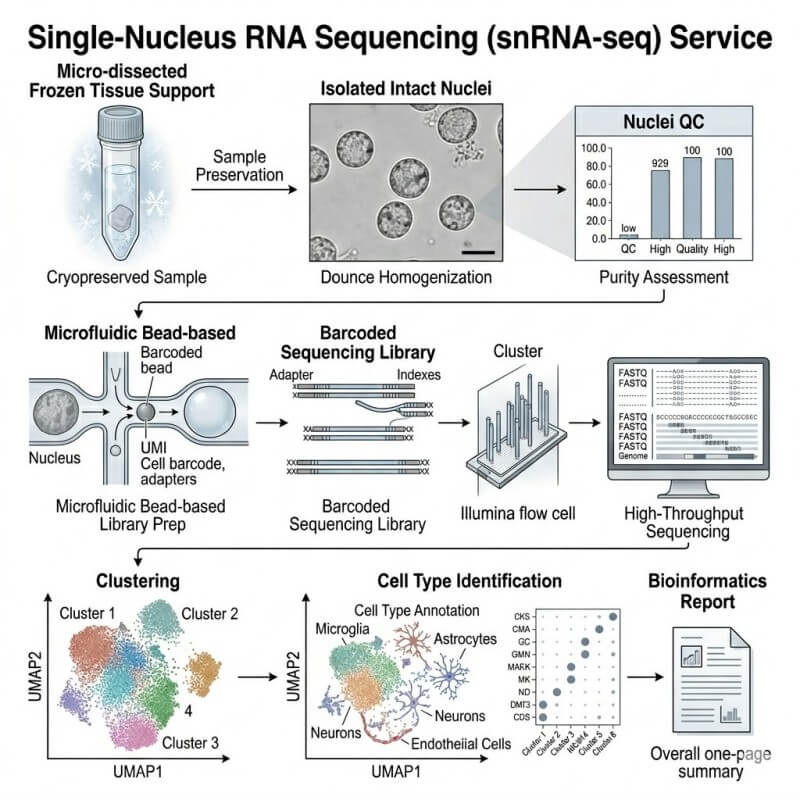
Our snRNA-seq workflow follows the sample from project intake through final data delivery. We combine technical sample handling with service-level review, so quality is checked before the data are used for biological interpretation.
A typical project moves through five stages: sample feasibility review, nuclei preparation or nuclei input assessment, library construction, sequencing, and bioinformatics analysis.
Project Intake and Sample Feasibility Review
We begin by reviewing your sample type, preservation method, tissue source, experimental groups, and downstream analysis goals.
Is the sample fresh, frozen, fixed, or already processed?
Is the tissue fragile, fatty, fibrotic, or hard to dissociate?
Are nuclei likely to be recoverable at useful quality?
Is the study design balanced across groups?
Are biological replicates labeled clearly?
Does the project need basic clustering only, or deeper interpretation?
This review helps us decide whether snRNA-seq is a reasonable fit and what sample preparation notes should be addressed before submission.
Nuclei Isolation or Input Assessment
Depending on the project scope, the workflow may begin from frozen tissue, fresh difficult tissue, or an isolated nuclei suspension.
For tissue-based projects, nuclei preparation generally includes tissue disruption, nuclear release, washing, debris reduction, filtration, counting, and quality review. For customer-prepared nuclei suspensions, we review input quality before moving into library preparation.
Nuclei concentration
Nuclear integrity
Singlet nuclei rate
Debris level
Clumping or aggregates
Background RNA or free nucleic acids
Inhibitors that may affect reverse transcription
A clean nuclei suspension is important because poor input can carry forward into lower sensitivity, weak cluster separation, or difficult annotation.
Library Construction and Sequencing QC
After input assessment, nuclei are processed for single-nucleus transcriptome library construction. Barcoded libraries are generated so transcript reads can be assigned back to individual nuclei.
Library yield and fragment profile
Read quality
Barcode and molecular barcodes performance
Mapping behavior
Nuclei recovery
Gene detection patterns
Overall sequencing data quality
These checks help determine whether the dataset is ready for downstream clustering and interpretation.
Data QC Before Biological Interpretation
Before we interpret the biology, we review technical quality.
Typical data QC includes filtering low-quality nuclei, reviewing gene and molecular barcodes distributions, assessing mitochondrial or ribosomal signal where relevant, checking potential doublets, and evaluating whether clusters are driven by biological differences or technical noise.
Only after this QC step do we move into annotation, marker gene review, group comparison, and optional advanced analysis.
Sample Requirements for snRNA-seq Projects
Sample quality strongly affects single-nucleus RNA sequencing results. The table below gives practical reference values based on N2 Jenomics Lab Pvt. Ltd. sample guidance for single-cell-related projects and common nuclei workflow considerations. Final requirements may vary by tissue type, preservation method, nuclei preparation route, and project design.
Sample Type
Recommended Input
Minimum / Reference Amount
Shipping or Storage
Key QC Checkpoints
Notes
Fresh tissue for nuclei workflow
Tissue submitted for nuclei preparation review
Fresh tissue reference: ≥0.2 g
Fully submerged in pre-cooled tissue preservation solution at 2–8°C
Nuclei workflow may be preferred for liver parenchymal cells, mature adipocytes, muscle, heart, brain, or nerve tissue.
For broader submission guidance, please review our Sample Submission Guidelines. For single-cell-related sample handling details, see our Single Cell Sequencing Sample Preparation Guidelines.
Bioinformatics Built for Single-Nucleus Interpretation
A single-nucleus RNA sequencing project should not stop at a count matrix. You need to know what the clusters mean, how confidently they can be annotated, and which outputs your team can inspect, reuse, and build on.
N2 Jenomics Lab Pvt. Ltd. connects quality control, clustering, marker gene detection, cell type annotation, and biological comparison in one analysis workflow. When your study requires more than standard processing, we can add deeper analysis around your tissue type, sample groups, and research question.
Minimum Analysis Deliverables
Deliverable
What You Receive
Why It Matters
Raw sequencing data
FASTQ files
Enables future reprocessing and archiving.
Processed expression matrix
Feature-barcode matrix or equivalent output
Forms the base for downstream single-nucleus analysis.
QC summary
Sequencing and nuclei-level quality metrics
Helps evaluate whether the dataset is suitable for interpretation.
Filtering summary
Notes on retained and removed nuclei
Improves transparency around quality decisions.
Dimensionality reduction plots
UMAP or t-SNE visualizations
Shows cluster structure across nuclei.
Clustering results
Cluster assignments and metadata
Supports cell population discovery.
Marker gene tables
Cluster-level marker genes
Helps define and validate cell identities.
Cell type annotation table
Annotated clusters with supporting markers
Connects computational clusters to biological meaning.
Cell composition overview
Proportion summaries by sample or group
Supports comparison across conditions.
Analysis report
Methods, figures, tables, and interpretation notes
Gives your team a readable project summary.
Optional Advanced Analysis Modules
Differential expression by cluster or group
Pathway, GO, or KEGG enrichment
Batch effect assessment and correction
Subcluster analysis for selected cell populations
Pseudotime or trajectory analysis
Cell-cell communication inference
Integration with public datasets
Comparative analysis across treatment, genotype, tissue region, or developmental stage
Spatial or multi-omics follow-up planning
For related transcriptomic services, you may also review our Single-cell RNA Sequencing page.
Reusable Files and Parameter Transparency
We do not treat single-nucleus bioinformatics as a black box. When applicable, we can provide reusable analysis objects, processed tables, figure files, and parameter notes so your internal bioinformatics team can review the workflow.
Metadata tables
Cluster annotation tables
Marker gene tables
Differential expression tables
Figure exports
Analysis reports
Seurat or Scanpy-compatible objects when applicable
Pipeline notes and parameter summaries
Choosing snRNA-seq Against Related Transcriptomic Options
The best transcriptomic method depends on your sample, your biological question, and the resolution you need. We help you choose a practical route before you commit samples.
Works well when intact whole cells are hard to recover
Nuclear transcript profiles may differ from whole-cell profiles
Choose this when tissue state or dissociation bias is the main concern.
Single-cell RNA sequencing
Fresh, viable cells or high-quality cell suspensions
Whole-cell single-cell transcriptome
Captures broader whole-cell RNA signals
Requires viable, clean, dissociated cells
Choose this when fresh cell recovery is strong and whole-cell information is needed.
Bulk RNA-seq
Tissue, cells, or RNA extract
Sample-average expression
Efficient for global expression comparison
Masks cell-type heterogeneity
Choose this when cell-level resolution is not required.
Spatial transcriptomics
Tissue sections
Spatially resolved transcriptomic signal
Preserves tissue architecture
Resolution and transcript depth vary by platform
Choose this when cell location and tissue structure are central to the study.
Single-cell multi-omics
High-quality cells or nuclei depending on assay
Multi-layer single-cell signal
Links transcriptome with chromatin, immune repertoire, or other modalities
Requires careful experimental design and higher complexity
Choose this when one molecular layer is not enough to answer the question.
Practical Selection Rules
Choose snRNA-seq when your sample is frozen, difficult to dissociate, fragile, or likely to lose important cell populations during whole-cell preparation.
Choose single-cell RNA sequencing when you can reliably prepare high-viability single-cell suspensions and want whole-cell transcriptome information.
Choose bulk RNA-seq when your main question is gene expression change at the sample level, not cell-type-specific interpretation.
Choose spatial transcriptomics when the position of cells or transcripts inside tissue architecture is essential.
Choose a multi-omics extension when gene expression alone cannot explain regulatory mechanisms. For example, you can pair transcriptomic profiling with chromatin accessibility through our scATAC-seq Service, or explore immune repertoire questions through our scTCR/BCR-seq Service.
Why Work With N2 Jenomics Lab Pvt. Ltd. for snRNA-seq
A successful snRNA-seq project requires more than sequencing capacity. It needs sample judgment, careful QC, consistent project execution, and analysis outputs that your team can understand and reuse.
Sample-First Project Design: We begin with your sample and research goal. Before library construction, our team reviews sample type, preservation method, study groups, expected biological variation, and downstream analysis needs.
QC-Aware Sequencing Execution: We apply QC thinking across sample review, nuclei quality assessment, library QC, sequencing data QC, nuclei filtering, cluster-level review, and analysis report preparation.
Custom Bioinformatics for Research Questions: We can adapt the analysis plan around your study design, including group comparison, selected cluster review, pathway analysis, trajectory analysis, or integration with public data.
Clear Deliverables for Review and Reuse: You receive outputs that your team can inspect and reuse, including raw data, processed matrices, QC summaries, annotated tables, figure files, and analysis reports.
References
snCED-seq: high-fidelity cryogenic enzymatic dissociation of nuclei for single-nucleus RNA-seq of FFPE tissues
Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows
Isolation of Nuclei from Human Snap-frozen Liver Tissue for Single-nucleus RNA Sequencing
Compliance / Disclaimer
For Research Use Only (RUO). This service is not intended for clinical diagnosis, medical interpretation, patient management, treatment guidance, or Direct-to-Consumer genetic testing.
Demo Results: What You Can Expect to See
Demo results help you understand the types of outputs a snRNA-seq project can produce. The examples below describe common result categories without implying fixed outcomes for every dataset.
Cell Clustering and Cell Type Annotation
A UMAP or t-SNE plot is often used to visualize nuclei after normalization, dimensionality reduction, and clustering. Each point represents a nucleus, and clusters are annotated using known marker genes, dataset context, and research-specific biology.
Typical visual: UMAP plot colored by cluster and annotated cell type.
How we use it: To identify major cell populations and guide do
Marker Gene and Cell Composition Views
Marker gene dot plots, heatmaps, or violin plots help support cluster identity. Cell composition plots then show how annotated populations differ across groups, tissues, treatments, or time points.
Typical visual: Marker gene dot plot plus stacked bar chart of cell population proportions.
How we use it: To connect computational clustering with interpretable biological categories.
Condition Comparison and Pathway-Level Interpretation
For studies with experimental groups, we can compare gene expression patterns within selected clusters or annotated cell types. Optional pathway analysis can help summarize biological themes behind the differential expression results.
Typical visual: Differential expression heatmap, volcano plot, or pathway enrichment bar chart.
How we use it: To prioritize cell types, genes, and pathways for follow-up experiments.
Compliance / Disclaimer
For Research Use Only (RUO). This service is not intended for clinical diagnosis, medical interpretation, patient management, treatment guidance, or Direct-to-Consumer genetic testing.
Case Study: FFPE and Complex Tissue snRNA-seq Feasibility
Source:snCED-seq: high-fidelity cryogenic enzymatic dissociation of nuclei for single-nucleus RNA-seq of FFPE tissues
Background
Archived and fixed tissue collections are valuable for retrospective tissue biology studies, but they create a major technical challenge for single-nucleus transcriptomics. RNA cross-linking, tissue processing, and nuclear recovery can affect whether a sample yields nuclei that are useful for sequencing and downstream analysis.
The 2025 Nature Communications study introduced snCED-seq, a cryogenic enzymatic dissociation strategy designed to improve nuclei extraction from FFPE tissue for single-nucleus RNA sequencing. The study focused on whether nuclei-based transcriptomics could recover cell-type-level information from difficult processed samples.
Methods
The authors developed a cryogenic enzymatic dissociation workflow and applied it to mouse and human tissue contexts. They evaluated nuclei extraction, transcript recovery, contamination signals, gene detection, clustering, marker-based annotation, and cell heterogeneity analysis.
The study compared fixed and frozen tissue contexts and used single-nucleus RNA sequencing to examine whether major brain-cell types and tissue heterogeneity could be resolved after nuclei preparation.
Results
In Figure 4, the authors showed that snCED-seq distinguished major brain-cell types. The figure included UMAP-based clustering of mouse hippocampal single-nucleus RNA profiles, marker-based cell-type annotation, cell frequency summaries, and differential expression-related views. The study reported 11 major cell types, including excitatory neurons, inhibitory neurons, astrocytes, oligodendrocytes, oligodendrocyte progenitor cells, microglia, Cajal-Retzius cells, and choroid plexus cells.
The same paper also reported human FFPE lung tissue heterogeneity analysis in Figure 6, supporting the broader point that nuclei-based transcriptomic workflows can be used to explore cell populations in complex processed tissues.
Conclusion
This paper supports a practical message for snRNA-seq service planning: when whole-cell workflows are limited by tissue preservation or dissociation constraints, nuclei-based transcriptomics can still provide cell-type-level structure, marker gene evidence, and interpretable heterogeneity analysis. For service projects, this makes sample feasibility review, nuclei QC, and bioinformatics interpretation essential parts of the workflow.
Compliance / Disclaimer
For Research Use Only (RUO). This service is not intended for clinical diagnosis, medical interpretation, patient management, treatment guidance, or Direct-to-Consumer genetic testing.
FAQs About Single-Nucleus RNA Sequencing Service
What sample types are best suited for single-nucleus RNA sequencing?
snRNA-seq is often a good fit for frozen tissue, archived tissue, fragile cells, large-cell tissues, and samples that are difficult to dissociate into viable whole-cell suspensions. Brain, heart, muscle, adipose-rich tissue, fibrotic tissue, and some tumor tissues are common examples.
When should I choose snRNA-seq instead of single-cell RNA sequencing?
Choose snRNA-seq when preparing a high-quality whole-cell suspension is difficult or likely to introduce bias. Choose single-cell RNA sequencing when fresh, viable, dissociated cells are available and whole-cell transcriptome capture is important for your study.
Can frozen tissue be used for snRNA-seq?
Yes. Frozen tissue is one of the main reasons researchers consider nuclei-based profiling. Sample quality, tissue type, freezing history, and nuclei recovery potential still need to be reviewed before project setup.
What QC checks are important for nuclei suspensions?
Important QC checks include nuclei concentration, nuclei integrity, singlet rate, debris level, clumping, free nucleic acid background, and potential inhibitors. Downstream data QC may also review detected genes, molecular barcodes distribution, cluster quality, and doublet risk.
What data files and analysis outputs will I receive?
Typical deliverables include raw sequencing data, processed expression matrices, QC summaries, clustering results, marker gene tables, cell type annotation tables, cell composition summaries, figure files, and an analysis report. Reusable analysis objects and parameter notes may be provided when applicable.
Can N2 Jenomics Lab Pvt. Ltd. help with cell type annotation?
Yes. We can support marker gene review, cluster annotation, and cell population interpretation based on the sample type, species, available references, and your study design. Annotation depth depends on tissue complexity and reference knowledge.
Do you support custom downstream analysis?
Yes. Optional analysis can include differential expression, pathway enrichment, batch effect assessment, subcluster analysis, pseudotime analysis, cell-cell communication inference, public dataset integration, and customized reporting.
Can snRNA-seq be combined with spatial or other omics methods?
Yes. snRNA-seq can be combined with related methods when the study needs both cell-type-level transcriptomics and additional biological context. For example, spatial transcriptomics can help place cell populations back into tissue architecture, while chromatin accessibility analysis can support regulatory interpretation.
Compliance / Disclaimer
For Research Use Only (RUO). This service is not intended for clinical diagnosis, medical interpretation, patient management, treatment guidance, or Direct-to-Consumer genetic testing.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India