Microbial Single-Cell Sequencing Service
Home
> Microbial Single-Cell Sequencing Service
Microbial Single-Cell Sequencing Services: High-Throughput SAG & scRNA-seq via MobiNova®
Powered by MobiNova®, N2 Jenomics Lab Pvt. Ltd. delivers a microbial single cell sequencing service built for complex samples. This page covers microbial single cell genome sequencing (SAG) and microbial single cell RNA sequencing for research-use-only projects. You get cell-level resolution when bulk methods blur rare taxa and states.
What we help you solve
Metagenomes can miss key genomes in low-abundance, uncultured populations.
Community averages hide strain diversity and mobile element carriage.
Bulk RNA profiles can mask rare response states under stress conditions.
What we provide
Single-cell genome sequencing (SAG / single-microbe genome) for strain-resolved recovery.
Microbial single-cell transcriptomics to profile heterogeneous bacterial expression states.
Optional packages linking genomes, mobile elements, and interpretable analysis outputs.
Why researchers choose N2 Jenomics Lab Pvt. Ltd.
Ultra-high throughput single-cell processing powered by the MobiNova® platform.
Flexible study design with 1–4 samples per run and scalable cell capture, successfully applied to diverse sample types.
Precise single-cell control with high capture efficiency and low multiplet rates.
Reliable delivery supported by contamination monitoring and reproducible SOPs.
Microbial communities rarely behave as a single "average" organism.
They are mixtures of strains, rare taxa, and transient cellular states.
Standard metagenomics can still leave gaps in genome recovery.
Bulk RNA profiling can still hide small but important subpopulations.
Single-cell methods move you from inference to direct linkage.
You can connect genetic elements to the cell that carries them.
You can also see how cells diverge under the same condition.
That matters in ecology, microbiomes, and industrial microbiology research.
Peer-reviewed work shows why this resolution is valuable.
In Microbe-seq, researchers generated >20,000 microbial SAGs from one donor.
They recovered many genomes and resolved strain structure at scale. (Zheng et al., 2022)
Reviews also highlight how single-cell approaches expose low-abundance roles. (Lloréns-Rico et al., 2022)
Feature
Bulk Metagenomics
Microbial Single-Cell (SAG)
Data Source
Pooled community DNA
Individual isolated cells
Resolution
Population average
Strain-level resolution
Rare Taxa
Often lost in "noise"
High sensitivity for low-abundance
HGT/Mobile Elements
Statistical inference
Direct physical linkage to host
Status
Genomic potential only
Active expression (via scRNA-seq)
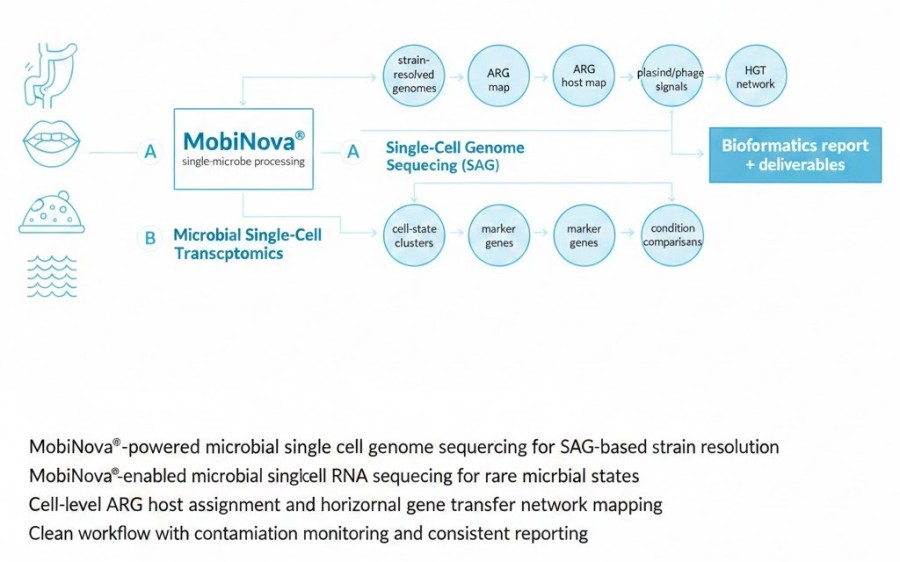
MobiNova® Platform Overview
MobiNova® gives our microbial single-cell sequencing service a scalable, research-grade foundation.
Across both genome and transcriptomics workflows, MobiNova® enables stable droplet generation, controlled single-cell partitioning, and standardised library construction for consistent downstream analysis.
MobiNova®-100: High-Throughput Single-Cell Library Preparation System
The MobiNova®-100 platform utilizes a patented water-in-oil droplet workflow to achieve superior single-cell partitioning. Unlike traditional methods, it is optimized for:
High Capture Efficiency: Maximizing the recovery of rare microbial cells from limited or complex biomass samples.
Ultra-Low Multiplet Rate: Precision microfluidic control ensures that each droplet contains a true single cell, minimizing data noise and cross-contamination.
Stable & Controlled Droplet Generation: Optimized instrument control allows for consistent co-encapsulation of cells with reagents and barcoded beads, ensuring reproducible library construction even for diverse microbial sizes and shapes.
MobiNova®-M1: Microbial Single-Cell Genome Sequencing Library Preparation System (Microbe-seq)
The MobiNova®-1 is purpose-built for the Microbe-seq workflow, offering a revolutionary leap beyond traditional metagenomics. By integrating sophisticated droplet microfluidics, it achieves:
Strain-Level Resolution Beyond MAGs: Unlike metagenomic binning, which often fails to distinguish closely related strains, MobiNova®-M1 recovers high-quality SAGs (Single-amplified Genomes) with precise strain-level specificity.
Direct Host-Phage/Plasmid Linkage: Provides the "missing link" by physically associating mobile genetic elements (MGEs) and antibiotic resistance genes (ARGs) with their specific bacterial hosts within a single droplet.
Broad Species Applicability: A species-agnostic platform that captures both cultured and "dark matter" (uncultured) microbes without PCR bias or GC-content limitations.
Seamless Complex Community Profiling: Optimized for high-complexity samples (gut, soil, marine), ensuring key organisms are assembled even at low abundance where bulk methods fail.
Choose the Right Modality: SAG, Transcriptomics, or Both
Choose microbial single-cell genome sequencing (SAG) when you need genomes
SAG is the best fit when your key question is "who is there."
It is also ideal when assembly gaps block your next step.
Common triggers include:
Metagenomes produce MAGs, but strain resolution is still weak.
Critical taxa are uncultured or of extremely low abundance.
You need a host-level assignment for plasmids, phages, or ARG context.
A high host background makes community sequencing less specific.
Choose microbial single-cell RNA sequencing when you need states
Single-cell transcriptomics is best for "what are they doing?"
It helps when heterogeneity is the real biology.
Common triggers include:
Bulk RNA-seq averages away rare but important expression programs.
You suspect bet-hedging, stress differentiation, or persistence-like states.
You need interpretable clusters, markers, and condition comparisons.
You tried bacterial scRNA-seq and hit feasibility barriers.
Choose a combined package when you need a complete story
Many projects benefit from a combined design.
Genomes explain capability and evolutionary context.
Transcriptomes explain active programmes under your conditions.
A combined plan can also improve interpretability for publication.
Quick guide to selecting microbial single-cell genome sequencing versus microbial single-cell transcriptomics.
Microbial Single-Cell Genome Sequencing Service
What SAG helps you answer
SAG gives you cell-resolved genome recovery.
It supports strain-resolved ecology and comparative genomics.
It is especially useful when MAGs cannot close the gaps.
With SAG, you can:
Recover genomes from uncultured or low-abundance microbes.
Separate closely related strains that co-exist in one sample.
Associate mobile genetic elements with a specific host genome.
Build higher-confidence datasets for pangenomes and phylogenomics.
Typical SAG projects we support
Environmental and marine microbiology
Ocean water, sediments, soils, and extreme environments.
Genome recovery for "uncultured" lineages and rare clades.
Microbiome and community genomics
Oral, gut, and mixed-community samples.
Strain-level genome sets for functional interpretation.
Platform centres and shared facilities
Batch submissions with consistent SOPs and stable reporting.
Emphasis on throughput, reproducibility, and cost control.
SAG deliverables
Your delivery can be data-only or interpretation-ready.
Typical SAG deliverables include:
Raw sequencing data (FASTQ) and run-level QC summary.
SAG assemblies and de-redundant genome sets, when applicable.
Genome quality metrics and contamination assessment report.
Functional annotation outputs suited for downstream analysis.
Optional joint package: SAG + metagenome co-interpretation.
Microbial Single-Cell Transcriptomics Sequencing Service
What bacterial single-cell transcriptomics reveals
Bacterial populations can split into distinct programmes.
Those differences can be rare and easy to miss.
Single-cell transcriptomics helps you detect and interpret them.
You can use microbial single-cell RNA sequencing to:
Identify rare stress and response programmes.
Quantify subpopulation shifts across conditions or timepoints.
Discover state-associated marker genes for validation.
Compare responses across strains or perturbations.
Typical transcriptomics projects we support
Antibiotic response and stress biology
Identify heterogeneous responses in the same culture.
Track state proportions across stress gradients.
Persistence and bet-hedging research
Capture rare survival-associated programmes.
Prioritise markers for targeted follow-up assays.
Biofilm and community behaviour
Resolve within-biofilm heterogeneity and specialisation.
Link state markers to environmental micro-niches.
Deliverables for technical readers and service buyers
We deliver results that can be interpreted and reused.
Typical outputs include:
Data QC summary and processing report.
Clustering and state definition with transparent rationale.
Marker gene lists per state and condition comparisons.
Pathway-level summaries and figure-ready plots.
Optional: state proportion shifts across conditions and timepoints.
Microbial single-cell RNA sequencing resolves heterogeneous bacterial states under stress.
End-to-End Workflow
The streamlined MobiNova® microbial single-cell sequencing workflow.
A typical project follows six clear steps:
Study design consultation and scope definition.
Sample intake review and feasibility checks.
MobiNova® single-microbe processing for your chosen modality.
Sequencing and primary data QC.
Bioinformatics, reporting, and interpretation alignment.
Delivery, handoff call, and optional iteration.
This workflow supports both single projects and batch pipelines.
It is designed for reproducible outputs across cohorts.
Contamination Control and Quality Reporting
SAG workflows are extremely sensitive to trace DNA.
This is why contamination monitoring is a buying criterion.
A strong service must be transparent about controls and QC.
Our reporting is designed for downstream confidence.
It helps you decide what to trust and how to use it.
Typical components include:
Process controls and documentation for clean handling.
Negative controls and interpretation of their read profiles.
Read-level and contig-level screening for likely contaminants.
Clear criteria for filtering and decontamination steps.
This focus aligns with community expectations for SAG projects.
It also supports reproducibility across batch submissions.
Contamination monitoring and transparent QC reporting for SAG projects.
Bioinformatics and Reporting Packages
Comprehensive bioinformatics workflow for microbial single-cell genomics. The process transitions from high-throughput cell partitioning and SAG generation to advanced downstream applications, including strain-level genome assembly, horizontal gene transfer (HGT) network analysis, and host-phage association mapping using the MobiNova® platform.
We structure deliverables for two audiences.
SAG analysis modules
Common modules include:
Assembly and genome set consolidation, when applicable.
Coverage and contiguity summaries for each recovered genome.
Completeness and contamination estimates with clear notes.
Functional annotation for downstream comparative workflows.
Optional integration: SAG + MAG joint analysis package.
Transcriptomics analysis modules
Common modules include:
QC summaries and filtering logic suitable for publication.
Clustering, marker selection, and condition comparisons.
State annotation grounded in your experimental context.
Microbiome samples: gut, oral, and mixed communities.
Cultures: pure isolates and defined consortia.
Intake questions that speed feasibility
These questions reduce risk and improve delivery quality:
Do you need SAG, transcriptomics, or both?
What sample type and expected complexity do you have?
Are targets uncultured or low-abundance?
Have you already metagenome data with unresolved gaps?
Is mobile element host assignment a key goal?
For transcriptomics, are stress or biofilm states central?
What deliverable depth do you need: data-only or interpretation?
What is your success metric for publication or downstream work?
Get Strain-Resolved Genomes and Interpretable Cell States
If metagenomics cannot close your genomes, SAG can help.
If bulk RNA hides rare states, single-cell transcriptomics can help.
With a MobiNova®-powered microbial single cell sequencing service, you can design a project that is both feasible and interpretable.
Contact our team to discuss your sample type and goals.
Get a free quote for SAG, transcriptomics, or a combined package.
Start your project with a structured intake checklist and control plan.
Research use only: N2 Jenomics Lab Pvt. Ltd. provides non-clinical research services only.
Trademark/Authorization Note
"MobiNova® is a registered trademark of its respective owner. N2 Jenomics Lab Pvt. Ltd. is an authorized user for the purposes of providing research-use-only services."
Microbial Single-Cell Seq Case Studies
Single-Cell Genome Sequencing Reveals Cell-Resolved Antibiotic Resistance Gene Flow in the Gut Microbiome
This 2025 study highlights the limitations of 'Gene Soup' in metagenomics and the necessity of SAGs for resolving ARG flow.
Reference
Ye L, Wu Y, Guo J, et al. Elucidation of population-based bacterial adaptation to antimicrobial treatment by single-cell sequencing analysis of the gut microbiome of a hospital patient. mSystems, 2025.
DOI: https://doi.org/10.1128/msystems.01631-24
1. Background
Limitations of Traditional Approaches and the Need for Single-Cell Resolution
Understanding how antibiotic resistance genes (ARGs) emerge and spread within complex microbial communities remains a major challenge in microbiome research. Traditionally, researchers have relied on bacterial culture and shotgun metagenomics. Culture-based methods are limited to a small fraction of cultivable organisms, while metagenomics provides only community-averaged genetic profiles.
As a result, metagenomic data often resemble a "gene soup":
ARGs can be detected, but their exact microbial hosts remain uncertain.
Low-abundance or uncultured organisms are easily overlooked.
Horizontal gene transfer (HGT) events cannot be directly assigned to specific cells.
Single-cell genome sequencing fundamentally changes this paradigm by enabling direct linkage between individual microbial cells and the genes they carry. In this study, single-cell amplified genomes (SAGs) were used to resolve ARG distribution, strain diversity, and gene flow within a human gut microbiome under antibiotic pressure.
Figure 1. Community composition and functional gene landscape revealed by SAGs
2. Methods
Single-Cell Genome Sequencing for Cell-Level ARG Attribution
To overcome the limitations of bulk approaches, the researchers applied microbial single-cell genome sequencing to gut samples collected during antibiotic treatment. Individual microbial cells were isolated and subjected to whole-genome amplification, generating thousands of single-cell amplified genomes (SAGs).
This approach enabled:
Genome recovery from uncultured and low-abundance microbes.
Direct assignment of ARGs to specific bacterial cells and strains.
Reconstruction of gene flow patterns across the microbial community.
By analysing SAG-derived genomes instead of pooled reads, the study achieved a one-to-one relationship between microbial cells and their genetic content—something not possible with conventional metagenomics.
Figure 2. Direct mapping of antibiotic resistance genes to individual bacterial hosts
3. Results
A Highly Connected Antibiotic Resistance Network at Single-Cell Resolution
3.1 Multi-Host ARGs and Cross-Species Co-Evolution
Analysis of SAGs revealed that the same ARG could be detected in phylogenetically distant bacterial hosts. For example, the aminoglycoside resistance gene aad9 appeared in both Bacteroidetes and Firmicutes, forming distinct evolutionary clusters.
This pattern indicates that ARGs are not static but actively evolve and diversify across multiple microbial hosts under antibiotic selection pressure.
Figure 3. Phylogenetic analysis of ARGs across multiple bacterial hosts
3.2 Horizontal Gene Transfer as a Community-Wide Process
The study identified 309 putative horizontal gene transfer events, involving not only ARGs but also genes linked to DNA repair, folate metabolism, and quorum sensing (e.g., folE, polA, queC).
These findings suggest that under antibiotic stress, microbes exchange both resistance genes and adaptation-enhancing genes, forming a highly interconnected ecological network that promotes community resilience.
Figure 4. Horizontal gene transfer network within the gut microbiome
3.3 Strain-Level Differentiation in a Key Pathogen
Focusing on Klebsiella pneumoniae, the researchers distinguished two coexisting strains (SC-KP1 and SC-KP2) that would likely be conflated in bulk analyses. SC-KP2 carried a broader repertoire of ARGs, including cfr(C) and fosXCC, and participated more actively in gene exchange with other gut microbes.
This strain-level resolution demonstrates how certain lineages may act as hubs for resistance gene dissemination.
Figure 5. Strain-level resolution of Klebsiella pneumoniae using SAGs
4. Conclusions
Implications for Microbiome and Resistance Research
This study demonstrates how microbial single-cell genome sequencing transforms resistance research from a population-level snapshot into a cell-resolved dynamic map. Key takeaways include:
The gut microbiome can rapidly evolve into a highly interconnected reservoir of ARGs under antibiotic pressure.
Single-cell approaches enable precise host assignment for resistance genes and mobile elements.
Strain-level resolution reveals heterogeneity even within clinically important species.
While this work is based on a single individual and requires validation in larger cohorts, it clearly illustrates the power of single-cell genomics for studying microbial adaptation and gene flow in complex communities.
Microbial Single-Cell Seq FAQs
Q: What is the difference between SAG and MAG?
A: MAGs are assembled from pooled community reads.
SAGs start from single microbes, enabling cell-resolved genomes.
SAG often helps when MAGs cannot resolve strains or gaps.
Q: Can you work with very complex samples?
A: Yes, complex samples are a common use case.
Feasibility depends on biomass, complexity, and host background.
We scope projects using an intake checklist and controls.
Q: How do you address contamination risk in SAG projects?
A: We use controls and provide transparent QC documentation.
We also report contamination screening and filtering rationale.
This supports downstream confidence and reproducibility.
Q: Do you deliver analysis or only sequencing data?
A: Both options are available.
Most teams choose an interpretation-ready report package.
Bioinformatics is especially valuable for single-cell datasets.
Q: Can I run a pilot first?
A: A pilot is often sensible for new sample types.
It helps set realistic success metrics and parameters.
It also reduces risk before scaling to batch submissions.
Q: How does microbial scRNA-seq handle the absence of poly-A tails in bacteria?
A: Unlike eukaryotic single-cell kits, our microbial transcriptomics workflow utilizes specialized r-RNA depletion and specific priming compatible with bacterial mRNA, ensuring high-fidelity capture of heterogeneous expression states.
Q: What is the recommended sample input for MobiNova® single-cell processing?
A: We typically require a cell concentration of 105 to 106 cells/mL in a specific buffer. Contact our team for a detailed project-specific intake checklist.
References:
High-throughput single-microbe genomics at strain resolution. (Zheng et al., 2022. DOI: https://doi.org/10.1126/science.abm1483)
Single-cell approaches for microbiome research and rare taxa. (Lloréns-Rico et al., 2022. DOI: https://doi.org/10.1016/j.cell.2022.06.040)
PETRI-seq, a scalable bacterial single-cell transcriptomics method. (Blattman et al., 2020. DOI: https://doi.org/10.1038/s41564-020-0729-6)
microSPLiT, split-pool barcoding for bacterial single-cell RNA-seq. (Kuchina et al., 2021. DOI: https://doi.org/10.1126/science.aba5257)
Niu, H., Gu, J. & Zhang, Y. Bacterial persisters: molecular mechanisms and therapeutic development.Sig Transduct Target Ther 9, 174 (2024) https://www.nature.com/articles/s41392-024-01866-5.
Blattman SB, Jiang W, McGarrigle ER, Liu M, Oikonomou P, Tavazoie S. Identification and genetic dissection of convergent persister cell states. Nature. 2024 Nov 6. https://www.nature.com/articles/s41586-024-08124-2
Ye L, Wu Y, Guo J, et al. Elucidation of population-based bacterial adaptation to antimicrobial treatment by single-cell sequencing analysis of the gut microbiome of a hospital patient.mSystems. 2025. doi:10.1128/msystems.01631-24
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India