Skim Sequencing Service: Low-Cost, High-Throughput Genotyping for Agriculture & Research
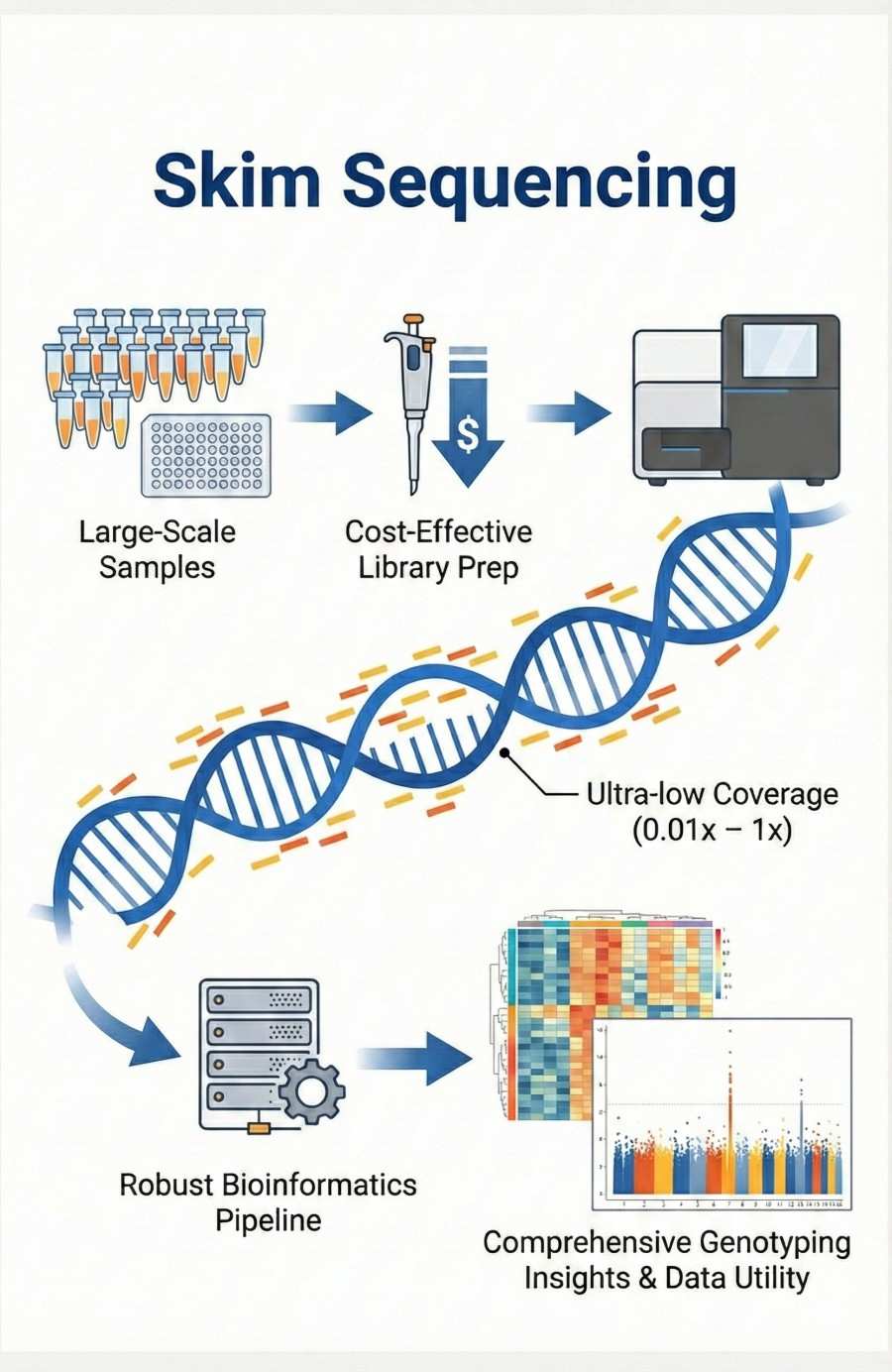
Skim Sequencing is an advanced genotyping service designed to revolutionize large-scale genetic studies by leveraging ultra-low coverage whole-genome sequencing (0.01x – 1x). This specific depth optimization allows us to drastically reduce sequencing costs while retaining the power to deliver comprehensive genetic insights. By combining cost-effective library preparation with robust bioinformatics pipelines, we enable the simultaneous analysis of thousands of samples without compromising on data utility. Whether for chromosome dosage estimation, translocation identification, or high-density marker discovery, our service makes whole-genome accessibility a reality for researchers and breeders operating on a budget.
Skim sequencing, also known as low-pass whole-genome sequencing, is a next-generation sequencing (NGS) application where each sample is sequenced at a shallow coverage (typically ranging from 0.01x to 1x). Unlike methods that sequence only a fraction of the genome (like GBS or RAD-seq), skim sequencing reads are randomly distributed across the entire genome. When combined with sophisticated imputation and analysis pipelines, this low-coverage data can be used to call variants, genotype individuals, and characterize genomic structures with high accuracy.
Originally developed to overcome the cost and scalability limitations of deep sequencing for large populations, it capitalizes on the ever-decreasing cost of sequencing to provide a "genome-wide" view that is both comprehensive and economical. Beyond high-abundance targets such as plastomes, these data can also be leveraged to identify low-copy nuclear genes, significantly expanding the scope of their applications.
Why Choose Skim Sequencing?
For agribusinesses, research institutions, and conservation programs, skim sequencing represents the optimal balance of data richness, throughput, and budget.
The primary driver for adoption is cost. Skim sequencing reduces the major bottleneck of library construction cost and time through highly multiplexed, low-volume protocols. Furthermore, it provides future-proof data. Unlike arrays, the raw sequence data can be re-analyzed as new genetic questions arise or reference genomes improve, protecting your investment.
Skim Sequencing Service Applications in Agricultural Genomics
Our service is specifically tailored to empower agriculture:
Genomic Selection & Accelerated Breeding: Rapidly genotype large breeding populations (e.g., thousands of lines) to predict complex trait performance and select superior parents, significantly shortening breeding cycles.
High-Resolution Genetic Mapping: Power genome-wide association studies (GWAS) and quantitative trait locus (QTL) mapping with dense, genome-wide markers to pinpoint genes controlling yield, disease resistance, drought tolerance, and quality traits.
Introgression & Backcross Tracking: Precisely identify and characterize desirable genomic segments (e.g., from wild relatives) in breeding lines. Monitor backcrossing progress to maintain essential genetics while adding new traits.
Structural & Cytogenetic Analysis: Detect chromosomal abnormalities, estimate copy number variation (dosage), and identify translocations or aneuploidy—critical for polyploid crops like wheat and for livestock health.
Genetic Diversity & Resource Characterization: Efficiently profile germplasm collections, heirloom varieties, and wild relatives to assess genetic diversity, identify unique alleles, and inform conservation strategies.
Skim Sequencing Service Workflow
Our streamlined, end-to-end workflow ensures reliability, quality, and rapid turnaround.
Skim Sequencing Bioinformatics Analysis
We transform raw sequencing data into actionable biological insights. Our standard and advanced packages include:
Standard Data Processing:
Demultiplexing & adapter trimming.
Quality control (FastQC, MultiQC).
Alignment to reference genome (BWA-MEM).
Raw variant calling (GATK).
Delivery of FASTQ, BAM, and initial VCF files.
Advanced Analysis Modules:
Genotype Imputation: Uses population-level haplotype information to accurately predict missing genotypes, effectively increasing the resolution and utility of low-coverage data.
Population Genetics: Analysis of genetic diversity, population structure, and phylogenetic relationships using tools like PLINK and ADMIXTURE.
Genome-Wide Association Study (GWAS): Identification of markers statistically associated with traits of interest.
Structural Variant Detection: Identification of copy number variants (CNVs), inversions, and translocations using read-depth and split-read analyses.
Assembly-Free Applications: For metagenomic or uncharacterized samples, we offer tools like Skmer, which computes genomic distances for identification without needing a reference genome.
For personalized bioinformatics analysis or specific research needs, please reach out to our experts for professional advice and support tailored to your project's requirements.
Sample Requirements for Skim Sequencing
To ensure project success, we recommend the following:
Sample Type
Minimum Quantity
Quality Metrics
Genomic DNA
100 ng (for library prep)
A260/A280: 1.8-2.0; A260/A230 >2.0. Intact on gel (high molecular weight).
Plant Tissue
Young leaf tissue (100-200 mg)
Fresh, frozen (in liquid N₂), or preserved in reliable buffer (e.g., CTAB, silica gel).
Animal Tissue
25 mg (e.g., ear notch, blood, semen)
Fresh-frozen or preserved in ethanol. Avoid cross-contamination.
Why Choose N2 Jenomics Lab Pvt. Ltd. for Skim Sequencing?
We are not just a service provider; we are a partner in your scientific discovery.
Proven Expertise & Technology: Our protocol is based on peer-reviewed, published methods developed for large-scale plant and animal genotyping. We implement state-of-the-art imputation pipelines proven in government-contracted work.
Scalability for Large-Scale Projects: Whether you are processing 100 or 10,000 samples, our optimized multiplexing workflow delivers consistent precision without exceeding your budget. We are fully equipped to support the extensive trials essential to modern agricultural research.
End-to-End Support & Customization: From experimental design to final interpretation, our team of PhD-level scientists provides continuous support. We customize analyses for diverse species—from row crops and livestock to specialty species and microbial communities.
Data Security & Compliance: We adhere to strict data confidentiality agreements. Our bioinformatics pipeline can optionally integrate with biosecurity screening tools (e.g., NCBI BLAST, SecureDNA) to screen sequences against pathogens of concern, ensuring responsible research conduct.
References:
Adhikari L., et al. A high-throughput skim-sequencing approach for genotyping, dosage estimation and identifying translocations. Sci Rep 12(1), 17583 (2022).
Kumar P., et al. Skim sequencing: an advanced NGS technology for crop improvement. J Genet 100, 38 (2021).
Sarmashghi S., et al. Skmer: assembly-free and alignment-free sample identification using genome skims. Genome Biol 20, 34 (2019).
Berger B.A., et al. The unexpected depths of genome-skimming data: A case study examining Goodeniaceae floral symmetry genes. Appl Plant Sci 5(10), 1700042 (2017).
FAQ
1. How low of coverage can I use, and what is the accuracy?
For well-characterized species with good reference genomes and population data for imputation, coverage as low as 0.1x to 0.5x can yield highly accurate genotype calls (e.g., for GWAS and genomic selection). For detecting structural variants or working with novel species, 1x or higher coverage may be recommended. Our bioinformaticians will advise on the optimal coverage for your goals.
2. My organism doesn't have a perfect reference genome. Can I still use skim sequencing?
Absolutely. For genetics within a population, a draft genome or a genome from a close relative is often sufficient for alignment and variant calling. Furthermore, assembly-free methods like Skmer can be used for sample identification and diversity analysis without any reference genome.
3. How does skim sequencing compare to whole-genome sequencing (WGS) for my breeding program?
Skim sequencing is essentially low-coverage WGS. The key difference is cost per sample. For the price of deep-sequencing (30x) one individual, you can skim-sequence hundreds. If your primary goals are genotyping, selection, and mapping—not discovering every single rare variant in an individual—skim sequencing provides far greater power and return on investment for breeding.
4. Can you handle samples from the field, like leaves stored in RNA-later or silica gel?
Yes. We have extensive experience processing diverse sample types. While high-quality, fresh-frozen DNA is ideal, we offer consultation on the best preservation methods for your field conditions and can perform extraction services if needed.
Case Study
Limited haplotype diversity underlies polygenic trait architecture across 70 years of wheat breeding
Population: NIAB Diverse MAGIC (500+ Recombinant Inbred Lines).
Method: Skim Sequencing (Low-coverage WGS) at ~0.3x coverage.
Outcome: 1.1 million high-quality SNPs imputed with >99% accuracy.
Key Takeaway: Skim sequencing provided 3x the call rate of direct genotyping and enabled high-resolution mapping of polygenic traits.
The Challenge: Genotyping a Complex Polyploid
Researchers at NIAB and UCL sought to analyze the genetic architecture of historical phenotypic changes in wheat. They created a Multi-parent Advanced Generation Intercross (MAGIC) population derived from 16 historical UK wheat varieties released between 1935 and 2004.
The Obstacles:
Genome Size: The wheat genome is massive (17 Gb) and hexaploid, making deep sequencing prohibitively expensive for hundreds of lines.
Array Limitations: Traditional SNP arrays often suffer from ascertainment bias (detecting only common variants) and struggle with polyploid complexity.
Scale: The study required genotyping over 500 recombinant inbred lines (RILs) to map traits effectively.
The Solution: Skim Sequencing + Imputation
Instead of using expensive deep sequencing or restrictive arrays, the team utilized Skim Sequencing. They sequenced the 550 RILs at an average depth of just 0.304x.
To recover high-quality genotype data from this sparse raw data, they applied imputation using STITCH software. This process leveraged the haplotype blocks inherited from the founders to fill in the gaps.
The Methodology
Founders: The 16 founder lines were sequenced more deeply using promoter-gene capture to create a haplotype reference.
Progeny: The 500+ RILs were sequenced at low coverage (~0.3x).
Imputation: Genotypes were inferred based on the probability of carrying specific founder haplotypes.
Validation: Accuracy Comparable to "Gold Standard" Arrays
The study validated the Skim Sequencing data against a subset of markers from the Axiom 35k SNP array. The results confirmed that low-coverage sequencing is highly reliable.
Metric
Result
Imputation Accuracy
99.1% concordance with array genotypes.
SNP Yield
1.13 million high-quality SNPs (vs. ~20k on the array).
Effective Call Rate
99.6% (increased 3-fold from raw read data).
Critical Insight: Downsampling analysis showed that genotypes could be accurately inferred from coverage as low as 0.076x per sample. Furthermore, imputation accuracy remained high (>98%) even without using the founders as a reference panel, demonstrating the method's robustness.
The high-density data generated via Skim Sequencing allowed researchers to dissect complex agronomic traits with precision.
1. High-Resolution QTL Mapping
The team mapped 136 genome-wide significant associations across 47 traits.
They identified 42 distinct genetic loci controlling traits like yield, disease resistance, and height.
Most traits were found to be highly polygenic, controlled by fine-scale shuffling of haplotypes.
2. Uncovering Trade-offs
The data revealed extensive pleiotropy (single genes affecting multiple traits). Specifically, they analyzed the negative trade-off between Grain Yield (GY) and Grain Protein Content (GPC).
Using the dense genotype data, they identified that the presence of awns (beards on the wheat ear) is associated with a positive deviation from the yield-protein trade-off.
This insight provides a clear target for breeders to simultaneously improve yield and protein.
3. Genomic Prediction
Using LASSO models on the Skim Seq data, the researchers achieved high prediction accuracy for out-of-sample lines.
Prediction accuracy averaged 0.43, using roughly 155 SNPs per phenotype.
The study concluded that future genetic gains would require selecting for dozens of polygenic alleles of small effect, facilitated by this type of genomic data.
Address:Registered Office: 150, Patparganj Industrial Area, New Delhi – 110092, India
Operational Address:National Institute of Plant Genome Research (BRIC - NGGF)
Lab No. 206 and 207, Aruna Asaf Ali Marg, P.O. Box No. 10531,
New Delhi – 110067, India